
ULTOMIRIS 1 100 mg-11 mL, solution à diluer pour perfusion IV, boîte de 1 flacon de 11 ml
Dernière révision : 04/07/2024
Taux de TVA : 2.1%
Laboratoire exploitant : ALEXION PHARMA FRANCE SAS
Source :

Hémoglobinurie paroxystique nocturne (HPN)
Ultomiris est indiqué pour le traitement de l'HPN chez les patients adultes et chez les patients pédiatriques pesant 10 kg ou plus :
- qui présentent une hémolyse avec un ou des symptômes cliniques indiquant une forte activité de la maladie ;
- qui sont stables sur le plan clinique après un traitement par l'eculizumab pendant au moins les 6 derniers mois.
Syndrome hémolytique et urémique atypique (SHUa)
Ultomiris est indiqué pour le traitement du SHUa chez les patients adultes et chez les patients pédiatriques pesant 10 kg ou plus, naïfs d'inhibiteur du complément ou ayant reçu un traitement par l'eculizumab pendant au moins 3 mois et présentant des signes de réponse à l'eculizumab.
Myasthénie acquise généralisée (MAg)
Ultomiris est indiqué en association au traitement standard pour le traitement de la MAg chez les patients adultes présentant des anticorps anti-récepteurs de l'acétylcholine (aRAch).
Maladie du spectre de la neuromyélite optique (NMOSD)
Ultomiris est indiqué pour le traitement de la NMOSD chez les patients adultes présentant des anticorps anti-aquaporine 4 (AQP4) (voir rubrique Propriétés pharmacodynamiques).
- Hypersensibilité à la substance active ou à l'un des excipients mentionnés à la rubrique Liste des excipients.
- Patients présentant une infection par Neisseria meningitidis non résolue lors de l'instauration du traitement (voir rubrique Mises en garde spéciales et précautions d'emploi).
- Patients sans vaccination à jour contre Neisseria meningitidis à moins qu'ils ne reçoivent une antibioprophylaxie appropriée jusqu'à 2 semaines après vaccination (voir rubrique Mises en garde spéciales et précautions d'emploi).
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administrés doivent être clairement enregistrés.
Infection à méningocoque grave
Du fait de son mécanisme d'action, l'utilisation du ravulizumab augmente la prédisposition du patient à une infection/une septicémie à méningocoque (Neisseria meningitidis). Une infection à méningocoque de tout sérogroupe peut survenir (voir rubrique Effets indésirables). Pour réduire le risque d'infection, tous les patients doivent être vaccinés contre les infections à méningocoque au moins deux semaines avant l'instauration du traitement par le ravulizumab, à moins que le risque dû au fait de retarder le traitement par ravulizumab soit supérieur à celui de développer une infection à méningocoque. Les patients pour lesquels le traitement par le ravulizumab a été initié dans un délai inférieur à deux semaines après la vaccination antiméningococcique doivent recevoir une antibioprophylaxie appropriée jusqu'à deux semaines après la vaccination. Les vaccins contre les sérogroupes A, C, Y, W135 et B lorsque disponibles, sont recommandés dans la prévention contre les sérogroupes méningococciques couramment pathogènes. Les patients doivent recevoir la primovaccination ou la vaccination de rappel conformément aux recommandations vaccinales nationales en vigueur. En cas de relais de l'eculizumab, le médecin doit s'assurer que la vaccination antiméningococcique du patient est à jour conformément aux recommandations vaccinales nationales en vigueur.
La vaccination peut ne pas suffire à éviter une infection à méningocoque. Les recommandations officielles relatives à l'utilisation appropriée des agents antibactériens doivent être prises en compte. Des cas d'infections/de septicémie à méningocoque graves ou d'évolution fatale ont été rapportés chez des patients traités par le ravulizumab et chez des patients traités par d'autres inhibiteurs de la voie terminale du complément. Tous les patients doivent être surveillés afin que tout signe précoce d'infection et de septicémie à méningocoque puisse être détecté, examinés immédiatement en cas de suspicion d'infection et traités par des antibiotiques appropriés si nécessaire. Les patients doivent être informés des signes et symptômes, ainsi que de la conduite à tenir pour obtenir une prise en charge médicale immédiate. Les médecins doivent remettre aux patients la brochure d'information patient et la carte patient.
Vaccination
Avant l'instauration du traitement par le ravulizumab, il est recommandé que les patients soient vaccinés conformément aux recommandations vaccinales en vigueur.
La vaccination peut suractiver le complément. Par conséquent, les patients atteints de maladies médiées par le complément peuvent présenter une augmentation des signes et des symptômes de leur pathologie sous-jacente. Les patients doivent donc être étroitement suivis, après avoir reçu la vaccination recommandée, afin que les symptômes de leur maladie puissent être surveillés.
Les patients âgés de moins de 18 ans doivent être vaccinés contre Haemophilus influenzae et contre les infections à pneumocoque, et respecter scrupuleusement les recommandations vaccinales nationales en vigueur pour chaque tranche d'âge.
Autres infections systémiques
Le
traitement par le ravulizumab doit être administré
avec précaution chez les patients présentant des infections systémiques
actives. Le ravulizumab inhibe l'activation de la
voie terminale du complément ; par conséquent, les patients peuvent présenter
une prédisposition accrue aux infections causées par Neisseria
sp. et par des bactéries encapsulées. Des cas
d'infections graves à Neisseriasp. (autres que Neisseria
meningitidis), notamment des infections
gonococciques disséminées, ont été rapportées.
Les
patients doivent être informés des mentions figurant dans la notice
d'information afin d'être sensibilisés à la possibilité d'infections
potentiellement graves ainsi qu'à leurs signes et symptômes. Les médecins
doivent informer les patients des mesures de prévention de la gonorrhée.
Réactions liées à la perfusion
L'administration du ravulizumab peut entraîner des réactions systémiques liées à la perfusion et des réactions allergiques ou d'hypersensibilité, y compris une anaphylaxie (voir rubrique Effets indésirables).
En cas de réaction liée à la perfusion systémique avec apparition de signes d'instabilité cardiovasculaire ou de détresse respiratoire, l'administration de ravulizumab doit être arrêtée et des mesures de support appropriées doivent être mises en place.
Interruption du traitement chez les patients atteints d'HPN
Si les patients atteints d'HPN interrompent le traitement par le ravulizumab, ils doivent être étroitement suivis afin que tout signe ou symptôme d'hémolyse intravasculaire grave puisse être détecté ; celle-ci est mise en évidence par une élévation du taux sérique de LDH (lactate déshydrogénase) associée à une diminution soudaine de la taille du clone HPN ou du taux d'hémoglobine ou par la réapparition de symptômes tels que : fatigue, hémoglobinurie, douleurs abdominales, difficultés respiratoires (dyspnée), événement indésirable vasculaire majeur (incluant thromboses), dysphagie, ou troubles de l'érection. La surveillance de tout patient interrompant le traitement par le ravulizumab doit se poursuivre pendant au moins 16 semaines pour détecter toute hémolyse et toute autre réaction. En cas d'apparition de signes et de symptômes d'hémolyse après l'interruption, y compris une élévation du taux de LDH, la reprise du traitement par le ravulizumab doit être envisagée.
Interruption du traitement chez les patients atteints de SHUa
Il
n'existe pas de données spécifiques concernant l'interruption du traitement par
le ravulizumab. Dans une étude observationnelle prospective
à long terme, l'interruption du traitement par inhibiteur de la protéine C5 du
complément (eculizumab) a entraîné un taux 13,5 fois
supérieur de récidive de MAT et il a été observé une tendance à la diminution
de la fonction rénale par rapport aux patients ayant poursuivi le traitement.
Si
les patients doivent interrompre le traitement par le ravulizumab,
ils doivent être étroitement suivis de manière continue afin que tout signe ou
symptôme de MAT puisse être détecté. Toutefois, le suivi peut s'avérer
insuffisant pour prédire ou prévenir des complications de MAT sévères.
Après
l'interruption du traitement, les complications de MAT peuvent être identifiées
si l'un des critères suivants est observé :
- au moins deux des résultats d'analyses biologiques suivants observés simultanément : diminution du nombre de plaquettes de 25 % ou plus par rapport à la valeur avant traitement ou par rapport au nombre de plaquettes le plus élevé durant le traitement par le ravulizumab ; augmentation du taux de créatinine sérique de 25 % ou plus par rapport à la valeur avant traitement ou au nadir durant le traitement par le ravulizumab ; ou augmentation du taux de LDH sérique de 25 % ou plus par rapport à la valeur avant traitement ou au nadir durant le traitement par le ravulizumab (les résultats doivent être confirmés par une deuxième mesure) ;
- l'un des symptômes de MAT suivants : modifications de l'état mental ou convulsions ou autres manifestations extrarénales de MAT incluant : anomalies cardiovasculaires, péricardite, symptômes gastro-intestinaux/diarrhée ; ou thrombose.
En cas de survenue de complications de MAT après l'interruption du traitement par le ravulizumab, il convient d'envisager la reprise du traitement par le ravulizumab, en commençant par la dose de charge et la dose d'entretien (voir rubrique Posologie et mode d'administration).
Interruption du traitement chez les patients atteints de MAg
La MAg étant une maladie chronique, les patients bénéficiant du traitement par le ravulizumab qui interrompent le traitement doivent être surveillés afin que les symptômes de la maladie sous-jacente puissent être détectés. En cas de survenue de symptômes de la MAg après l'interruption du traitement, la reprise du traitement par le ravulizumab doit être envisagée.
Interruption du traitement chez les patients atteints de NMOSD
La NMOSD étant une maladie chronique, les patients bénéficiant du traitement par le ravulizumab qui interrompent le traitement doivent être surveillés afin que les symptômes de poussée de la NMOSD puissent être détectés. En cas de survenue de symptômes de poussée de la NMOSD après l'interruption du traitement, la reprise du traitement par le ravulizumab doit être envisagée.
Relais de l'eculizumab par le ravulizumab
Le traitement par le ravulizumab n'est pas recommandé chez les patients atteints de MAg qui ne répondent pas au schéma posologique autorisé de l'eculizumab.
Teneur en sodium
Ultomiris 300 mg/3 mL et 1 100 mg/11 mL solutions à diluer pour perfusion
Après dilution avec une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %), ce médicament contient 0,18 g de sodium par volume de 72 mL à la dose maximale, ce qui équivaut à 9,1 % de l'apport alimentaire quotidien maximal recommandé par l'OMS de 2 g de sodium pour un adulte.
Ultomiris 300 mg/30 mL solution à diluer pour perfusion
Après dilution avec une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %), ce médicament contient 2,65 g de sodium par volume de 720 mL à la dose maximale, ce qui équivaut à 133 % de l'apport quotidien maximal recommandé par l'OMS de 2 g de sodium pour un adulte.
Résumé du profil de sécurité
Les effets indésirables les plus fréquents observés avec le ravulizumab sont : céphalées (28,2 %), infection des voies respiratoires supérieures (19,9 %), rhinopharyngite (19,5 %), diarrhée (16,9 %), pyrexie (16,4 %), nausées (13,7 %), arthralgie (13,2 %), fatigue (13,1 %), dorsalgie (12,6 %), douleurs abdominales (11,8 %) et vertiges (10,1 %). Les effets indésirables les plus graves sont les infections à méningocoque (0,7 %), incluant septicémies à méningocoque, encéphalites méningococciques et infections à méningocoque (voir rubrique Mises en garde spéciales et précautions d'emploi) et l'infection gonococcique disséminée (0,1 %).
Tableau des effets indésirables
Le tableau 9 présente les effets indésirables rapportés dans les études cliniques et depuis la commercialisation .
Les effets indésirables sont présentés par classe de systèmes d'organes
MedDRA et fréquence selon la convention suivante : très fréquent (≥
1/10) ; fréquent (≥ 1/100, < 1/10) ; peu fréquent (≥ 1/1 000, <
1/100) ; rare (≥ 1/10 000, < 1/1 000) ; très rare (< 1/10 000) et
fréquence indéterminée (ne peut être estimée sur la base des données
disponibles).
Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 9 : Effets indésirables rapportés dans les études cliniques et depuis la commercialisation
| Classe de systèmes d'organes MedDRA | Très fréquent (≥ 1/10) | Fréquent (≥ 1/100, < 1/10) | Peu fréquent (≥ 1/1 000, < 1/100) |
| Infections et infestations | Infection des voies aériennes supérieures, rhinopharyngite | Infection du tractus urinaire | Infection à méningocoquea, infection gonococcique disséminéeb |
| Affections du système immunitaire | Hypersensibilitéd | Réaction anaphylactiquec | |
| Affections du système nerveux | Céphalées, vertiges | ||
| Affections gastro-intestinales | Diarrhée, nausées, douleurs abdominales | Vomissements, dyspepsie | |
| Affections de la peau et du tissu sous-cutané | Urticaire, prurit, rash | ||
| Affections musculosquelettiques et du tissu conjonctif | Arthralgie, dorsalgie | Myalgie, contractures musculaires | |
| Troubles généraux et anomalies au site d'administration | Pyrexie, fatigue | Syndrome pseudo-grippal, frissons, asthénie | |
| Lésions, intoxications et complications liées aux procédures | Réaction liée à la perfusion |
a Le terme « infection à méningocoque » inclut les termes préférentiels « infection à méningocoque », « septicémie à méningocoque » et « encéphalite méningococcique ».
b Le terme « infection gonococcique disséminée » inclut les termes préférentiels « infection gonococcique disséminée » et « infection gonococcique ».
c Fréquence estimée sur la base des données depuis la commercialisation.
d Le terme « hypersensibilité » regroupe les termes préférentiels « hypersensibilité médicamenteuse avec lien de causalité » et « hypersensibilité ».
b Le terme « infection gonococcique disséminée » inclut les termes préférentiels « infection gonococcique disséminée » et « infection gonococcique ».
c Fréquence estimée sur la base des données depuis la commercialisation.
d Le terme « hypersensibilité » regroupe les termes préférentiels « hypersensibilité médicamenteuse avec lien de causalité » et « hypersensibilité ».
Description de certains effets indésirables
Infection/septicémie/encéphalite à méningocoque
La vaccination réduit, mais n'élimine pas, le risque d'infections à
méningocoque. Dans les études cliniques, moins de 1 % des patients ont
développé une infection à méningocoque grave pendant le traitement par
ravulizumab ; tous étaient des adultes atteints d'HPN ou de NMOSD et
étaient vaccinés. Voir la rubrique Mises en garde spéciales et précautions d'emploi
pour les informations sur la prévention et le traitement lors d'une
suspicion d'infection à méningocoque. Chez les patients traités par
ravulizumab, les infections à méningocoque se sont présentées sous
forme de septicémie à méningocoque ou d'encéphalite méningococcique.
Les patients doivent être informés des signes et symptômes d'infection
à méningocoque et avertis de la nécessité de consulter immédiatement un
médecin.
Réactions à la perfusion
Les réactions à la perfusion ont été fréquentes (≥ 1 %) dans les études
cliniques. Ces événements, qui étaient d'intensité légère à modérée et
transitoires, comprenaient : dorsalgie, douleurs abdominales,
contractures musculaires, chute de la pression artérielle, augmentation
de la pression artérielle, frissons, gêne dans un membre,
hypersensibilité (réaction allergique), dysgueusie (goût désagréable)
et somnolence. Ces réactions n'ont pas nécessité l'arrêt du traitement
par le ravulizumab.
Immunogénicité
Dans les études réalisées chez des patients adultes atteints d'HPN (n =
475), dans une étude chez des patients pédiatriques atteints d'HPN (N =
13), dans les études chez des patients atteints de SHUa (n = 89), dans
une étude chez des patients atteints de MAg (n = 86) et dans une étude
chez des patients atteints de NMOSD (n = 58), 2 patients (0,3 %) ont
développé des anticorps anti-médicament au cours du traitement par le
ravulizumab (un patient adulte atteint d'HPN et un patient adulte
atteint de SHUa). Ces anticorps anti-médicament étaient de nature
transitoire, de faible titre et n'étaient pas corrélés à la réponse
clinique ou aux événements indésirables.
Population pédiatrique
Hémoglobinurie paroxystique nocturne (HPN)
Chez les patients atteints d'HPN (N = 13, âgés de 9 ans à 17 ans)
inclus dans l'étude pédiatrique menée dans l'HPN (ALXN1210-PNH-304), le
profil de sécurité du ravulizumab s'est révélé similaire à celui
observé chez les patients adultes atteints d'HPN. Les effets
indésirables les plus fréquemment rapportés chez les patients
pédiatriques atteints d'HPN étaient des douleurs abdominales, des
nausées, une rhinopharyngite et des céphalées, survenues chez trois
patients (23,1 %).
Syndrome d'anémie hémolytique et urémique atypique (SHUa)
Chez les patients présentant un SHUa (N = 34, âgés de 10 mois à moins
de 18 ans) inclus dans l'étude ALXN1210-aHUS-312, le profil de sécurité
du ravulizumab s'est révélé similaire à celui observé chez les patients
adultes présentant des signes de SHUa. Les profils de sécurité dans les
différentes tranches d'âge de la population pédiatrique se sont avérés
similaires. Les données de sécurité chez les patients âgés de moins de
2 ans sont limitées à quatre patients. L'effet indésirable le plus
fréquemment rapporté chez les patients pédiatriques était la fièvre
(32,3 %).
Myasthénie acquise généralisée (MAg)
Le ravulizumab n'a pas été étudié chez les patients pédiatriques atteints de MAg.
Maladie du spectre de la neuromyélite optique (NMOSD)
Le ravulizumab n'a pas été étudié chez les patients pédiatriques atteints de NMOSD.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés après autorisation du médicament est importante. Elle permet une surveillance continue du rapport bénéfice/risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration - voir Annexe V.
AVANT INSTAURATION DU TRAITEMENT
:
- Vérifier que les patients aient été vaccinés contre Neisseria
meningitidis au moins 2 semaines avant l'initiation du traitement. Si le délai
est inférieur à 2 semaines entre la vaccination et l'instauration du traitement,
donner une antibioprophylaxie appropriée pendant les 2 semaines suivant la
vaccination.
- Vaccination de tous les patients conformément aux
recommandations vaccinales en vigueur.
Les patients âgés de moins de 18 ans
doivent être vaccinés contre les infections à Haemophilus influenzae et à
pneumocoque.
Après avoir reçu la vaccination recommandée, les patients
doivent être étroitement suivis pour surveiller les symptômes de leur
maladie.
SURVEILLANCE DU TRAITEMENT :
- Surveiller tout signe précoce
d'infection à méningocoque, examiner immédiatement en cas de suspicion
d'infection et traiter par une antibiothérapie appropriée si nécessaire.
INFORMER LE PATIENT :
- des mentions figurant dans la notice afin
d'être sensibilisé à la possibilité d'infections potentiellement graves ainsi
qu'à leurs signes et symptômes ;
- des mesures de prévention de la
gonorrhée.
SURVEILLANCE APRES INTERRUPTION DU TRAITEMENT :
- Chez
les patients atteints d'HPN, poursuivre la surveillance pendant au moins 16
semaines afin de détecter tout signe d'hémolyse intravasculaire grave mise en
évidence par une élévation du taux sérique de LDH (lactate déshydrogénase)
associée à une diminution soudaine de la taille du clone HPN ou du taux
d'hémoglobine ou par la réapparition de symptômes tels que : fatigue,
hémoglobinurie, douleur abdominale, difficultés respiratoires (dyspnée),
évènement indésirable vasculaire majeur (incluant thromboses), dysphagie, ou
troubles de l'érection.
- Chez les patients atteints de SHU atypique,
surveiller tout signe de micro-angiopathie thrombotique de manière continue. Le
suivi pouvant s'avérer insuffisant pour prédire ou prévenir des complications de
MAT sévères, celles-ci peuvent être identifiées si l'un des critères suivants
est observé :
. au moins deux des résultats d'analyses biologiques suivants
observés simultanément :
diminution du nombre de plaquettes de 25 % ou plus
par rapport à la valeur avant traitement ou par rapport au nombre de plaquettes
le plus élevé durant le traitement par le ravulizumab ;
augmentation du taux
de créatinine sérique de 25 % ou plus par rapport à la valeur de référence ou au
nadir durant le traitement par le ravulizumab ; ou augmentation du taux de LDH
sérique de 25 % ou plus par rapport à la valeur avant traitement ou au nadir
durant le traitement par le ravulizumab (les résultats doivent être confirmés
par une deuxième mesure) ;
. l'un des symptômes de MAT suivants :
modifications de l'état mental ou convulsions ou autres manifestations
extrarénales de MAT, notamment : anomalies cardiovasculaires, péricardite,
symptômes gastro-intestinaux / diarrhée ; ou thrombose.
- Chez les patients
atteints de MAg ou de NMOSD, surveiller afin de
détecter les symptômes de la maladie sous-jacente. En cas de survenue de
symptômes de la MAg ou
de NMOSD après l'interruption du traitement, la reprise du traitement par
le ravulizumab doit être
envisagée.
Pour les patients traités concomitamment par échange
plasmatique (EP), plasmaphérèse (PP) ou immunoglobulines intraveineuses, une
dose supplémentaire de ravulizumab est nécessaire.
Population pédiatrique
:
- Le ravulizumab n'a pas été étudié chez les patients pédiatriques atteints
d'HPN dont le poids est inférieur à 30 kg. Chez ces patients, la posologie du
ravulizumab est basée sur la posologie utilisée chez les patients pédiatriques
atteints de SHUa, selon les données pharmacocinétiques/pharmacodynamiques
(PK/PD) disponibles chez les patients atteints de SHUa ou d'HPN traités par le
ravulizumab.
- Le ravulizumab n'a pas été étudié chez les patients
pédiatriques atteints de MAg ou de NMOSD.
INFORMER :
- Les femmes en âge de procréer d'utiliser une contraception efficace pendant le traitement et jusqu'à 8 mois après l'arrêt du traitement.
- Les femmes allaitantes d'interrompre l'allaitement pendant le traitement et jusqu'à 8 mois après la fin du traitement.
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et jusqu'à 8 mois après l'arrêt du traitement.
Grossesse
Il n'existe pas de donnée clinique sur l'utilisation du ravulizumab chez la femme enceinte.
Il n'a pas été réalisé d'étude préclinique de toxicité sur la reproduction avec le ravulizumab (voir la rubrique Données de sécurité préclinique).
Des études de toxicité sur la reproduction ont été réalisées chez la
souris avec la molécule murine analogue BB5.1 afin d'évaluer les effets
de l'inhibition de la protéine C5 sur le système reproducteur. Il n'a
pas été identifié de toxicité spécifique du produit sur la
reproduction dans ces études. Dans la mesure où les IgG humaines
traversent la barrière placentaire humaine, le ravulizumab est
susceptible de provoquer une inhibition de la voie terminale du
complément au niveau de la circulation fœtale.
Les études effectuées chez l'animal sont insuffisantes pour permettre
de conclure sur la toxicité sur la reproduction (voir rubrique Données de sécurité préclinique).
Chez les femmes enceintes, l'utilisation du ravulizumab peut être envisagée après une évaluation du rapport bénéfice/risque.
Allaitement
On ne sait pas si le ravulizumab est excrété dans le lait maternel. Lors des études précliniques de toxicité sur la reproduction réalisées chez la souris avec la molécule murine analogue BB5.1, il n'a pas été identifié d'effets indésirables chez les petits suite à la consommation du lait de mères traitées.
Un risque pour les nourrissons ne peut être exclu.
Dans la mesure où de nombreux médicaments et immunoglobulines sont
excrétés dans le lait humain et en raison du risque d'effets
indésirables graves chez les nourrissons allaités, l'allaitement doit
être interrompu pendant le traitement par le ravulizumab et jusqu'à 8
mois après la fin du traitement.
Fertilité
Aucune étude préclinique spécifique des effets du ravulizumab sur la fertilité n'a été réalisée.
Dans les études précliniques de toxicité sur la reproduction réalisées
chez la souris avec la molécule murine analogue BB5.1, il n'a pas été
identifié d'effets indésirables sur la fertilité des femelles ou des
mâles traités.
Aucune étude d'interaction n'a été réalisée. Étant donné l'effet inhibiteur potentiel du ravulizumab sur la cytotoxicité dépendante du complément induite par le rituximab, le ravulizumab peut diminuer les effets pharmacodynamiques attendus du rituximab.
Un traitement chronique par immunoglobulines humaines intraveineuses (IgIV) peut interférer avec le mécanisme de recyclage des anticorps monoclonaux tels que le ravulizumab induit par le récepteur Fc endosomal néonatal (FcRn) et donc diminuer les concentrations sériques du ravulizumab.
Voir la rubrique Posologie et mode d'administration pour les recommandations en cas de traitement concomitant par EP, PP ou IgIV.
Le ravulizumab doit être administré par un professionnel de santé et sous la surveillance d'un médecin ayant l'expérience de la prise en charge des patients atteints de troubles hématologiques, rénaux, neuromusculaires ou neuro-inflammatoires.
Posologie
Patients
adultes atteints d'HPN, de SHUa, de MAg ou de NMOSD
Le schéma
posologique recommandé consiste en une dose de charge suivie de doses
d'entretien, administrées par perfusion intraveineuse. Les doses à administrer
sont basées sur le poids du patient, comme indiqué dans le tableau 1. Chez les
patients adultes (≥ 18 ans), les doses d'entretien doivent être
administrées une fois toutes les 8 semaines, en commençant deux semaines après
l'administration de la dose de charge.
Le schéma d'administration peut occasionnellement varier de ± 7 jours par rapport au jour de perfusion prévu (hormis pour la première dose d'entretien de ravulizumab), mais la dose suivante doit être administrée conformément au schéma initial.
Tableau 1 :
Schéma posologique du ravulizumab en fonction du
poids chez les patients adultes pesant 40 kg ou plus
| Poids (kg) | Dose de charge (mg) | Dose d'entretien (mg)* | Intervalle posologique |
| ≥ 40 à < 60 | 2 400 | 3 000 | Toutes les 8 semaines |
| ≥ 60 à < 100 | 2 700 | 3 300 | Toutes les 8 semaines |
| ≥ 100 | 3 000 | 3 600 | Toutes les 8 semaines |
* La première
dose d'entretien est administrée deux semaines après la dose de charge.
Les instructions pour l'instauration du traitement chez les patients naïfs d'inhibiteur du complément ou en cas de relais de l'eculizumab sont présentées dans le tableau 2.
Tableau 2 : Instructions pour l'instauration du traitement
par le ravulizumab
| Population | Dose de charge de ravulizumab en fonction du poids | Moment de la première dose d'entretien de ravulizumab en fonction du poids |
| Patients n'étant pas sous traitement par le ravulizumab ou l'eculizumab | Lors de l'instauration du traitement | 2 semaines après la dose de charge de ravulizumab |
| Patients sous traitement par l'eculizumab | Lors de la prochaine dose d'eculizumab planifiée | 2 semaines après la dose de charge de ravulizumab |
Patients pédiatriques atteints d'HPN ou de SHUa
Patients pédiatriques dont le poids est ≥ 40 kg
Ces patients doivent être traités conformément au schéma posologique recommandé chez l'adulte (tableau 1).
Patients pédiatriques dont le poids est ≥ 10 kg et < 40 kg
Pour les
patients pédiatriques dont le poids est compris entre ≥ 10 kg et < 40
kg, les posologies en fonction du poids et les intervalles posologiques sont
présentés dans le tableau 3.
En cas de
relais de l'eculizumab par le ravulizumab,
la dose de charge de ravulizumab doit être
administrée deux semaines après la dernière perfusion d'eculizumab
; les doses d'entretien sont ensuite administrées selon le schéma posologique
en fonction du poids indiqué dans le tableau 3, en commençant deux semaines
après l'administration de la dose de charge.
| Poids (kg) | Dose de charge (mg) | Dose d'entretien (mg)* | Intervalle posologique |
| ≥ 10 à < 20 | 600 | 600 | Toutes les 4 semaines |
| ≥ 20 à < 30 | 900 | 2 100 | Toutes les 8 semaines |
| ≥ 30 à < 40 | 1 200 | 2 700 | Toutes les 8 semaines |
* La première
dose d'entretien est administrée 2 semaines après la dose de charge.
Le ravulizumab n'a pas été étudié chez les patients pédiatriques atteints d'HPN dont le poids est inférieur à 30 kg. Chez ces patients, la posologie recommandée est basée sur la posologie utilisée chez les patients pédiatriques atteints de SHUa, sur la base des données pharmacocinétiques/pharmacodynamiques (PK/PD) disponibles chez les patients atteints de SHUa ou d'HPN traités par le ravulizumab.
L'HPN est une maladie chronique et il est recommandé de poursuivre le traitement par le ravulizumab durant toute la vie du patient, à moins que l'interruption du traitement par le ravulizumab ne soit cliniquement justifiée (voir rubrique Mises en garde spéciales et précautions d'emploi).
Dans le SHUa, un traitement par le ravulizumab visant à faire disparaître les manifestations de microangiopathie thrombotique (MAT) doit se poursuivre pendant une durée minimale de 6 mois. Au-delà de cette période, la durée du traitement doit être envisagée individuellement pour chaque patient. Les patients présentant un risque plus élevé de récidive de MAT selon l'avis du médecin en charge du patient (ou si cela s'avère cliniquement justifié) peuvent nécessiter un traitement chronique (voir rubrique Mises en garde spéciales et précautions d'emploi).
Chez les patients adultes atteints de MAg ou de NMOSD, le traitement par le ravulizumab n'a été étudié que dans le cadre d'une administration chronique (voir rubrique Mises en garde spéciales et précautions d'emploi).
Le ravulizumab n'a pas été étudié chez les patients atteints de MAg de classe V de la MGFA (Myasthenia Gravis Foundation of America).
Administration
de doses supplémentaires après un traitement par échange plasmatique (EP),
plasmaphérèse (PP) ou immunoglobulines intraveineuses (IgIV)
Il a été
démontré que le traitement par échange plasmatique (EP), plasmaphérèse (PP) ou
immunoglobulines intraveineuses (IgIV) diminue les
concentrations sériques du ravulizumab. Une dose
supplémentaire de ravulizumab est nécessaire en cas
de traitement par EP, PP ou IgIV (tableau 4).
Tableau 4 :
Dose supplémentaire de ravulizumab après un
traitement par EP, PP ou IgIV
| Poids (kg) | Dose de ravulizumab la plus récente (mg) | Dose supplémentaire (mg) après chaque séance d'EP ou de PP | Dose supplémentaire (mg) après la fin d'un cycle de traitement par IgIV |
| ≥ 40 à < 60 | 2 400 | 1 200 | 600 |
| 3 000 | 1 500 | ||
| ≥ 60 à < 100 | 2 700 | 1 500 | 600 |
| 3 300 | 1 800 | ||
| ≥ 100 | 3 000 | 1 500 | 600 |
| 3 600 | 1 800 | ||
| Moment d'administration de la dose supplémentaire de ravulizumab | Dans les 4 heures suivant chaque séance d'EP ou de PP | Dans les 4 heures suivant lafin d'un cycle de traitement par IgIV | |
Abréviations :
IgIV = immunoglobulines intraveineuses, kg =
kilogrammes, EP = échange plasmatique, PP = plasmaphérèse.
Populations particulières
Sujets âgés
Aucun
ajustement de la dose n'est nécessaire chez les patients atteints d'HPN, de SHUa, de MAg ou de NMOSD âgés de
65 ans et plus. Aucun élément n'indique que des précautions particulières
soient nécessaires lors de l'administration du traitement à une population
gériatrique, bien que l'expérience avec le ravulizumab
dans les études cliniques chez les patients âgés atteints d'HPN, de SHUa ou de NMOSD soit limitée.
Insuffisance
rénale
Aucun
ajustement de la dose n'est nécessaire chez les patients présentant une
insuffisance rénale (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
La
sécurité et l'efficacité du ravulizumab n'ont pas été
étudiées chez les patients présentant une insuffisance hépatique ; toutefois,
les données pharmacocinétiques semblent indiquer qu'aucun ajustement de la dose
n'est nécessaire chez les patients présentant une insuffisance hépatique.
Population pédiatrique
La sécurité et l'efficacité du ravulizumab chez les enfants atteints d'HPN ou de SHUa pesant moins de 10 kg n'ont pas été établies. Les données actuellement disponibles sont décrites à la rubrique Effets indésirables mais aucune recommandation sur la posologie ne peut être donnée.
La sécurité et l'efficacité du ravulizumab chez les enfants atteints de MAg ou de NMOSD n'ont pas été établies. Aucune donnée n'est disponible.
Mode d'administration
Perfusion
intraveineuse uniquement.
Le médicament
doit être administré à l'aide d'un filtre de 0,2 µm et ne doit pas être
administré en injection intraveineuse directe ou en bolus.
Ultomiris 300 mg/30 mL solution à diluer pour perfusion ne doit pas être mélangé avec Ultomiris 300 mg/3 mL ou 1 100 mg/11 mL solutions à diluer pour perfusion.
Ultomiris 300 mg/3 mL et 1 100 mg/11 mL solutions à diluer pour perfusion
Ultomiris solution à diluer pour perfusion est présenté en flacons de 3 mL et 11 mL (100 mg/mL) et doit être dilué pour obtenir une concentration finale de 50 mg/mL. Après dilution, Ultomiris doit être administré par perfusion intraveineuse en utilisant une pompe de type pousse-seringue ou une pompe à perfusion pendant une durée minimale de 0,17 à 1,3 heure (soit 10 à 75 minutes) en fonction du poids (voir le tableau 5 et le tableau 6 ci-dessous).
Tableau 5 :
Débit d'administration des doses pour Ultomiris 300
mg/3 mL et 1 100 mg/11 mL
solutions à diluer pour perfusion
| Poids (kg)a |
Dose de charge (mg) |
Durée minimale de perfusion
Minutes (heures) |
Dose d'entretien (mg) |
Durée minimale de perfusion
Minutes (heures) |
| ≥ 10 à < 20b | 600 | 45 (0,8) | 600 | 45 (0,8) |
| ≥ 20 à < 30b | 900 | 35 (0,6) | 2 100 | 75 (1,3) |
| ≥ 30 à < 40b | 1 200 | 31 (0,5) | 2 700 | 65 (1,1) |
| ≥ 40 à < 60 | 2 400 | 45 (0,8) | 3 000 | 55 (0,9) |
| ≥ 60 à < 100 | 2 700 | 35 (0,6) | 3 300 | 40 (0,7) |
| ≥ 100 | 3 000 | 25 (0,4) | 3 600 | 30 (0,5) |
a Poids au moment du
traitement.
Tableau 6 :
Débit d'administration des doses supplémentaires d'Ultomiris
300 mg/3 mL et 1 100 mg/11 mL
solutions à diluer pour perfusion
| Poids (kg)a |
Dose supplémentaireb (mg) |
Durée minimale de perfusion
Minutes (heure) |
| ≥ 40 à < 60 | 600 | 15 (0,25) |
| 1 200 | 25 (0,42) | |
| 1 500 | 30 (0,5) | |
| ≥ 60 à < 100 | 600 | 12 (0,20) |
| 1 500 | 22 (0,36) | |
| 1 800 | 25 (0,42) | |
| ≥ 100 | 600 | 10 (0,17) |
| 1 500 | 15 (0,25) | |
| 1 800 | 17 (0,28) |
a Poids au moment du
traitement.
Ultomiris 300 mg/30 mL solution à diluer pour perfusion
Ultomiris solution à diluer pour perfusion est présenté en flacon de 30 mL (10 mg/mL) et doit être dilué à une concentration finale de 5 mg/mL. Après dilution, Ultomiris doit être administré par perfusion intraveineuse en utilisant une pompe de type pousse-seringue ou une pompe à perfusion pendant une durée minimale de 0,4 à 3,3 heures (soit 22 à 194 minutes) en fonction du poids (voir le tableau 7 et le tableau 8 ci-dessous).
Tableau 7 :
Débit d'administration des doses pour Ultomiris 300
mg/30 mL solution à diluer pour perfusion
| Poids (kg)a |
Dose de charge (mg) |
Durée minimale de perfusion
Minutes (heures) |
Dose d'entretien (mg) |
Durée minimale de perfusion
Minutes (heures) |
| ≥ 10 à < 20b | 600 | 113 (1,9) | 600 | 113 (1,9) |
| ≥ 20 à < 30b | 900 | 86 (1,5) | 2 100 | 194 (3,3) |
| ≥ 30 à < 40b | 1 200 | 77 (1,3) | 2 700 | 167 (2,8) |
| ≥ 40 à < 60 | 2 400 | 114 (1,9) | 3 000 | 140 (2,3) |
| ≥ 60 à < 100 | 2 700 | 102 (1,7) | 3 300 | 120 (2,0) |
| ≥ 100 | 3 000 | 108 (1,8) | 3 600 | 132 (2,2) |
a Poids au moment du
traitement.
Tableau 8 : Débit d'administration des doses supplémentaires d'Ultomiris 300 mg/30 mL solution à diluer pour perfusion
| Poids (kg)a |
Dose supplémentaireb (mg) |
Durée minimale de perfusion
Minutes (heure) |
| ≥ 40 à < 60 | 600 | 30 (0,5) |
| 1 200 | 60 (1,0) | |
| 1 500 | 72 (1,2) | |
| ≥ 60 à < 100 | 600 | 23 (0,4) |
| 1 500 | 60 (1,0) | |
| 1 800 | 65 (1,1) | |
| ≥ 100 | 600 | 22 (0,4) |
| 1 500 | 60 (1,0) | |
| 1 800 | 65 (1,1) |
a Poids au moment du
traitement.
Pour les instructions concernant la dilution du médicament avant administration, voir la rubrique Précautions particulières d'élimination et de manipulation.
Durée de conservation :
18 mois.
Après dilution, le médicament doit être utilisé immédiatement. Toutefois, la stabilité physico-chimique du produit dilué a été démontrée pendant une durée allant jusqu'à 24 heures à une température comprise entre 2 °C et 8 °C et jusqu'à 4 heures à température ambiante.
Précautions particulières de conservation :
À conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Conserver le flacon dans l'emballage extérieur à l'abri de la lumière.
Pour les conditions de conservation du médicament après dilution, voir la rubrique Durée de conservation.
Ce médicament ne doit pas être mélangé avec d'autres médicaments à l'exception de ceux mentionnés dans la rubrique Précautions particulières d'élimination et de manipulation.
La dilution ne doit être réalisée qu'en utilisant comme diluant une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %).
En cas de surdosage, la perfusion doit être arrêtée immédiatement, et le patient doit être étroitement surveillé afin que des signes ou symptômes d'effets indésirables puissent être détectés et un traitement symptomatique approprié doit être instauré.
Classe pharmacothérapeutique : Immunosuppresseurs, immunosuppresseurs sélectifs, Code ATC : L04AA43
Mécanisme d'action
Le ravulizumab est un anticorps monoclonal de type IgG2/4k qui se lie sélectivement à la protéine C5 du complément, ce qui inhibe son clivage en C5a (l'anaphylatoxine pro-inflammatoire) et C5b (la sous-unité d'initiation du complexe d'attaque membranaire [CAM ou C5b-9]) et empêche la génération du complexe C5b-9. Le ravulizumab préserve les composants précoces de l'activation du complément qui sont essentiels à l'opsonisation des microorganismes et à l'élimination des complexes immuns.
Effets pharmacodynamiques
Après
l'administration de ravulizumab chez des patients adultes
et pédiatriques atteints d'HPN naïfs d'inhibiteur du complément ou
préalablement traités par l'eculizumab dans les
études de phase III, il a été observé à la fin de la première perfusion une
inhibition immédiate, complète et maintenue de la protéine C5 libre sérique
(concentration < 0,5 µg/mL), qui a persisté
pendant toute la période de traitement de 26 semaines chez tous les patients.
Une inhibition immédiate et complète de la protéine C5 sérique libre a
également été observée chez les patients adultes et pédiatriques atteints de SHUa, chez les patients adultes atteints de MAg et chez les patients adultes atteints de NMOSD à la fin
de la première perfusion et pendant toute la période de traitement principale.
L'ampleur et la durée de la réponse pharmacodynamique chez les patients
atteints d'HPN, de SHUa, de MAg
ou de NMOSD dépendaient de l'exposition au ravulizumab.
Des taux de protéine C5 libre inférieurs à 0,5 µg/mL
étaient corrélés à un contrôle maximal de l'hémolyse intravasculaire et à une
inhibition complète de la voie terminale du complément. Dans la MAg, l'activation de la voie terminale du complément
entraîne un dépôt de CAM au niveau de la jonction neuromusculaire et une
altération de la transmission neuromusculaire. Dans la NMOSD, l'activation de
la voie terminale du complément provoque la formation du CAM et une
inflammation induite par la protéine C5a, une nécrose des astrocytes et une
atteinte des cellules gliales et des neurones environnants.
Efficacité et sécurité cliniques
Hémoglobinurie paroxystique nocturne (HPN)
La sécurité et l'efficacité du ravulizumab chez les patients adultes atteints d'HPN ont été évaluées au cours de deux études de phase III randomisées en ouvert, contrôlées contre comparateur actif :
- une étude menée chez des patients adultes atteints d'HPN naïfs d'inhibiteur du complément,
- une étude menée chez des patients adultes atteints d'HPN préalablement traités par l'eculizumab qui étaient stables sur le plan clinique après traitement par l'eculizumab depuis au moins 6 mois.
Le
ravulizumab était administré conformément à la
posologie recommandée à la rubrique Posologie et mode d'administration
(4 perfusions de ravulizumab sur 26 semaines), alors
que l'eculizumab était administré selon la posologie
autorisée de 600 mg par semaine pendant les 4 premières semaines et 900 mg
toutes les 2 semaines (15 perfusions sur 26 semaines).
Les patients avaient reçu une vaccination antiméningococcique
avant, ou lors de, l'instauration du traitement par le ravulizumab
ou l'eculizumab ou une antibioprophylaxie appropriée
jusqu'à 2 semaines après la vaccination.
Dans les deux études de phase III, il n'y avait aucune différence notable en
termes de caractéristiques démographiques ou initiales entre les groupes de
traitement par le ravulizumab et l'eculizumab. Les antécédents transfusionnels sur 12 mois
étaient similaires entre les groupes de traitement par le ravulizumab
et l'eculizumab dans chacune des études de phase III.
Étude chez des patients adultes atteints d'HPN naïfs d'inhibiteur du complément (ALXN1210-PNH-301)
L’étude chez des patients naïfs d’inhibiteur du complément était une étude de phase III multicentrique, randomisée en ouvert, contrôlée contre comparateur actif d’une durée de 26 semaines menée chez 246 patients naïfs de traitement par inhibiteur du complément avant l’inclusion dans l’étude, suivie d’une période d’extension à long terme au cours de laquelle tous les patients ont reçu le ravulizumab. Les patients éligibles à cette étude devaient présenter un niveau élevé d'activité de la maladie, défini par un taux de LDH ≥ 1,5 × la limite supérieure de la normale (LSN) lors de la sélection, associé à la présence dans les trois mois précédant la sélection d'un ou plusieurs des signes ou symptômes de l'HPN suivants : fatigue, hémoglobinurie, douleurs abdominales, difficultés respiratoires (dyspnée), anémie (hémoglobine < 10 g/dL), antécédents d'événement indésirable vasculaire majeur (incluant thrombose), dysphagie ou troubles de l'érection ou antécédents de transfusion de globules rouges en raison de l'HPN.
Plus de 80 % des patients des deux groupes de traitements avaient des antécédents de transfusion au cours des 12 mois précédant l'inclusion dans l'étude. La majorité de la population de patients naïfs d'inhibiteur du complément de l'étude présentaient une hémolyse élevée lors de l'inclusion ; 86,2 % des patients inclus avaient un taux de LDH élevé ≥ 3 × LSN, ce qui est une mesure directe de l'hémolyse intravasculaire dans le cadre de l'HPN.
Le tableau 10 présente les caractéristiques initiales des patients atteints d'HPN inclus dans l'étude menée chez des patients naïfs d'inhibiteur du complément, sans différence cliniquement significative notable observée entre les groupes de traitement.
Tableau
10 : Caractéristiques initiales dans l'étude menée chez des patients naïfs
d'inhibiteur du complément
| Paramètre | Statistiques |
Ravulizumab
(n = 125) |
Eculizumab
(n = 121) |
| Âge (ans) lors du diagnostic d'HPN |
Moyenne
(ET) Médiane Min, max |
37,9 (14,90) 34,0 15, 81 |
39,6 (16,65) 36,5 13, 82 |
| Âge (ans) lors de la première perfusion dans l'étude |
Moyenne
(ET) Médiane Min, max |
44,8 (15,16) 43,0 18, 83 |
46,2 (16,24) 45,0 18, 86 |
| Sexe (n, %) |
Masculin
Féminin |
65 (52,0) 60 (48,0) |
69 (57,0) 52 (43,0) |
| Taux de LDH avant traitement |
Moyenne
(ET) Médiane |
1 633,5 (778,75) 1 513,5 |
1 578,3 (727,06) 1 445,0 |
|
Nombre
de patients ayant reçu des transfusions de concentré
érythrocytaire au cours des 12 mois précédant la première dose |
n (%) | 103 (82,4) | 100 (82,6) |
|
Unités
de concentré érythrocytaire transfusées
au
cours des 12 mois précédant la première dose |
Total
Moyenne (ET) Médiane |
925 9,0 (7,74) 6,0 |
861 8,6 (7,90) 6,0 |
| Taille totale du clone HPN érythrocytaire | Médiane | 33,6 | 34,2 |
| Taille totale du clone HPN granulocytaire | Médiane | 93,8 | 92,4 |
|
Patients
présentant des pathologies liées à l'HPNa
avant
le consentement éclairé Anémie Hématurie ou hémoglobinurie Anémie aplasique Insuffisance rénale Syndrome myélodysplasique Complications liées à la grossesse Autreb |
n (%) |
121 (96,8)
103 (82,4) 81 (64,8) 41 (32,8) 19 (15,2) 7 (5,6) 3 (2,4) 27 (21,6) |
120 (99,2)
105 (86,8) 75 (62,0) 38 (31,4) 11 (9,1) 6 (5,0) 4 (3,3) 13 (10,7) |
a D'après les antécédents médicaux.
b La catégorie « autre » indiquée sur le cahier d'observation incluait : thrombopénie, insuffisance rénale chronique, pancytopénie, ainsi qu'un certain nombre d'autres pathologies.
b La catégorie « autre » indiquée sur le cahier d'observation incluait : thrombopénie, insuffisance rénale chronique, pancytopénie, ainsi qu'un certain nombre d'autres pathologies.
Les co-critères principaux d'évaluation étaient l'absence de recours à la transfusion et l'hémolyse mesurée directement par la normalisation du taux de LDH (taux de LDH ≤ 1 × LSN ; la LSN du taux de LDH est de 246 U/L). Les principaux critères d'évaluation secondaires étaient la variation relative du taux de LDH par rapport à l'inclusion, la modification de la qualité de vie (questionnaire FACIT-Fatigue), le pourcentage de patients présentant un épisode hémolytique et le pourcentage de patients présentant une stabilisation de l'hémoglobine.
Le ravulizumab s'est avéré non inférieur à l'eculizumab pour les deux co-critères principaux d'évaluation, à savoir l'absence de recours à la transfusion de concentré érythrocytaire conformément aux recommandations du protocole et la normalisation du taux de LDH entre le jour 29 et le jour 183 et pour les quatre principaux critères d'évaluation secondaires (figure 1).
Figure 1 : Analyse des co-critères principaux d'évaluation et des critères d'évaluation secondaires - Population complète d'analyse (étude chez des patients naïfs d'inhibiteur du complément)

Note
: le triangle noir indique les marges de non-infériorité et les points gris
indiquent les estimations ponctuelles.
Abréviations : LDH = lactate déshydrogénase ; IC = intervalle de confiance ;
FACIT = Functional Assessment
of ChronicIllnessTherapy (évaluation
fonctionnelle dans le traitement des maladies chroniques).
L’analyse finale de l’efficacité a porté sur l’ensemble des patients ayant été traités par le ravulizumab (n = 244), chez lesquels la durée médiane de traitement était de 1 423 jours. L’analyse finale a confirmé que les réponses au traitement par le ravulizumab observées pendant la période d’évaluation principale étaient maintenues pendant toute la durée de l’étude.
Étude chez des patients adultes atteints d'HPN préalablement traités par l'eculizumab (ALXN1210-PNH-302)
L’étude chez des patients préalablement traités par l’eculizumab était une étude de phase III multicentrique, randomisée en ouvert, contrôlée contre comparateur actif d’une durée de 26 semaines menée chez 195 patients atteints d’HPN qui étaient stables sur le plan clinique (LDH ≤ 1,5 × LSN) après avoir été traités par l’eculizumab pendant au moins les 6 mois précédents, suivie d’une période d’extension à long terme au cours de laquelle tous les patients ont reçu le ravulizumab.
Les antécédents médicaux d'HPN étaient similaires entre les groupes de traitement par le ravulizumab et par l'eculizumab. Les antécédents transfusionnels sur 12 mois étaient similaires entre les groupes de traitement par le ravulizumab et par l'eculizumab et plus de 87 % des patients des deux groupes de traitement n'avaient pas reçu de transfusion dans les 12 mois précédant l'inclusion dans l'étude. La taille totale moyenne du clone HPN érythrocytaire était de 60,05 %, celle du clone HPN granulocytaire était de 83,30 % et celle du clone HPN monocytaire était de 85,86 %.
Le tableau 11 présente les caractéristiques initiales des patients atteints d'HPN inclus dans l'étude menée chez des patients préalablement traités par l'eculizumab, sans différence cliniquement significative notable observée entre les groupes de traitement.
Tableau 11 : Caractéristiques initiales dans l'étude chez des patients préalablement traités par l'eculizumab
| Paramètre | Statistiques |
Ravulizumab
(n = 97) |
Eculizumab
(n = 98) |
| Âge (ans) lors du diagnostic d'HPN | Moyenne (ET) | 34,1 (14,41) | 36,8 (14,14) |
| Médiane | 32,0 | 35,0 | |
| Min, max | 6, 73 | 11, 74 | |
| Âge (ans) lors de la première perfusion dans l'étude | Moyenne (ET) | 46,6 (14,41) | 48,8 (13,97) |
| Médiane | 45,0 | 49,0 | |
| Min, max | 18, 79 | 23, 77 | |
| Sexe (n, %) | Masculin | 50 (51,5) | 48 (49,0) |
| Féminin | 47 (48,5) | 50 (51,0) | |
| Taux de LDH avant le traitement | Moyenne (ET) | 228,0 (48,71) | 235,2 (49,71) |
| Médiane | 224,0 | 234,0 | |
|
Nombre
de patients ayant reçu des transfusions de concentré érythrocytaire/sang total au cours des 12 mois précédant la première dose |
n (%) | 13 (13,4) | 12 (12,2) |
|
Unités
de concentré érythrocytaire/sang total transfusées au cours des 12 mois précédant la première dose |
Total
Moyenne (ET) Médiane |
103 7,9 (8,78) 4,0 |
50 4,2 (3,83) 2,5 |
|
Patients
présentant des pathologies liées à l'HPNa avant le consentement éclairé Anémie Hématurie ou hémoglobinurie Anémie aplasique Insuffisance rénale Syndrome myélodysplasique Complications liées à la grossesse Autreb |
n (%) |
90 (92,8) 64 (66,0) 47 (48,5) 34 (35,1) 11 (11,3) 3 (3,1) 4 (4,1) 14 (14,4) |
96 (98,0) 67 (68,4) 48 (49,0) 39 (39,8) 7 (7,1) 6 (6,1) 9 (9,2) 14 (14,3) |
a
D'après les antécédents médicaux.
b La catégorie « autre » incluait thrombopénie, insuffisance rénale, pancytopénie, ainsi qu'un certain nombre d'autres pathologies.
b La catégorie « autre » incluait thrombopénie, insuffisance rénale, pancytopénie, ainsi qu'un certain nombre d'autres pathologies.
Le critère d'évaluation principal était l'hémolyse, mesurée par la variation relative du taux de LDH par rapport à l'inclusion. Les critères d'évaluation secondaires étaient le pourcentage de patients présentant un épisode hémolytique, la qualité de vie (questionnaire FACIT-Fatigue), l'absence de recours à la transfusion et le pourcentage de patients présentant une stabilisation de l'hémoglobine.
Le ravulizumab s'est avéré non inférieur à l'eculizumab pour le critère d'évaluation principal, à savoir la variation relative du taux de LDH entre l'inclusion et le jour 183, et pour les quatre principaux critères d'évaluation secondaires (figure 2).
Figure 2 : Analyse du critère d'évaluation principal et des critères d'évaluation secondaires - Population complète d'analyse (étude chez des patients préalablement traités par l'eculizumab)

Note
: le triangle noir indique les marges de non-infériorité et le point gris
indique les estimations ponctuelles.
Abréviations : LDH = lactate déshydrogénase ; IC = intervalle de confiance.
L'analyse finale de l'efficacité a porté sur l'ensemble des patients préalablement traités par le ravulizumab (n = 192), chez lesquels la durée médiane de traitement était de 968 jours. L'analyse finale a confirmé que les réponses au traitement par le ravulizumab observées pendant la période d'évaluation principale étaient maintenues pendant toute la durée de l'étude.
Syndrome hémolytique et urémique atypique (SHUa)
Étude chez les patients adultes atteints de SHUa (ALXN1210-aHUS-311)
L'étude
chez l'adulte était une étude de phase III multicentrique, à bras unique, menée
chez des patients atteints de SHUa documenté, naïfs
de traitement par inhibiteur du complément avant leur inclusion dans l'étude et
présentant des signes de microangiopathie
thrombotique (MAT). Après une période d'évaluation initiale de 26 semaines, les
patients pouvaient être inclus dans une phase d'extension de l'étude d'une durée
allant jusqu'à 4,5 ans.
Au total, 58 patients atteints de SHUa documenté ont
été inclus. Selon les critères d'inclusion, les patients présentant une MAT due
à un purpura thrombotique thrombocytopénique (PTT),
ou à un syndrome hémolytique et urémique à Escherichia coli producteur
de toxines Shiga (SHU à STEC) étaient exclus. Deux patients ont été exclus de
la population complète d'analyse en raison d'un diagnostic confirmé de SHU à
STEC. Quatre-vingt-treize pour cent des patients présentaient des signes (aux
niveaux cardiovasculaire, pulmonaire, du système nerveux central,
gastro-intestinal, cutané et musculo-squelettique) ou des symptômes extrarénaux
de SHUa lors de l'inclusion.
Le tableau 12 présente les caractéristiques démographiques et initiales des 56 patients adultes inclus dans l'étude ALXN1210-aHUS-311 qui ont constitué la population complète d'analyse.
Tableau 12 : Caractéristiques initiales dans l'étude menée chez des patients adultes
| Paramètre | Statistiques | Ravulizumab (N = 56) |
| Âge (ans) lors de la première perfusion |
Moyenne (ET) Min, max |
42,2 (14,98) 19,5, 76,6 |
|
Sexe
Masculin |
n (%) | 19 (33,9) |
|
Origine
ethnique Asiatique Caucasienne Autre |
n (%) |
15 (26,8) 29 (51,8) 12 (21,4) |
| Antécédents de greffe | n (%) | 8 (14,3) |
| Plaquettes (109/L), sang |
n Médiane (min, max) |
56 95,25 (18, 473) |
| Hémoglobine (g/L), sang |
n Médiane (min, max) |
56 85,00 (60,5, 140) |
| LDH (U/L), sérum |
n Médiane (min, max) |
56 508,00 (229,5, 3 249) |
| DFGe (mL/min/1,73 m2) |
n (%) Médiane (min, max) |
55 10,00 (4, 80) |
| Patients sous dialyse | N (%) | 29 (51,8) |
| Patientes en postpartum | N (%) | 8 (14,3) |
Note
: les pourcentages sont basés sur le nombre total de patients.
Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ; max = maximum ; min = minimum.
Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ; max = maximum ; min = minimum.
Le critère d'évaluation principal était la réponse complète de la MAT pendant la période d'évaluation initiale de 26 semaines, attestée par une normalisation des paramètres hématologiques (plaquettes ≥ 150 × 109/L et LDH ≤ 246 U/L) et une amélioration ≥ 25 % de la créatininémie par rapport à l'inclusion. Les patients devaient satisfaire à chaque critère de réponse complète de la MAT lors de deux évaluations distinctes réalisées à intervalle d'au moins quatre semaines (28 jours) et lors de toute mesure intermédiaire.
Une réponse complète de la MAT a été observée chez 30 des 56 patients (53,6 %) pendant la période d'évaluation initiale de 26 semaines, comme indiqué dans le tableau 13.
Tableau
13 : Analyse de la réponse complète de la MAT et des critères de la réponse
complète de la MAT pendant la période d'évaluation initiale de 26 semaines
(ALXN1210aHUS-311)
| Total | Répondeurs | ||
| n | Proportion (IC à 95 %)a | ||
| Réponse complète de la MAT | 56 | 30 | 0,536 (0,396 ; 0,675) |
|
Critère
de la réponse complète de la MAT Normalisation des plaquettes Normalisation de la LDH Amélioration ≥ 25 % de la créatininémie par rapport à l'inclusion |
56 56 56 |
47 43 33 |
0,839 (0,734 ; 0,944) 0,768 (0,648 ; 0,887) 0,589 (0,452 ; 0,727) |
| Normalisation hématologique | 56 | 41 | 0,732 (0,607 ; 0,857) |
a Les
IC à 95 % de la proportion étaient basés sur la méthode d'approximation
asymptotique gaussienne avec une correction de continuité.
Abréviations : IC = intervalle de confiance ; LDH = lactate déshydrogénase ; MAT = microangiopathie thrombotique.
Abréviations : IC = intervalle de confiance ; LDH = lactate déshydrogénase ; MAT = microangiopathie thrombotique.
Quatre patients supplémentaires ont présenté une réponse complète de la MAT, qui a été confirmée après la période d'évaluation initiale de 26 semaines (avec une réponse complète de la MAT survenant aux jours 169, 302, 401 et 407), entraînant une réponse complète de la MAT globale chez 34 des 56 patients (60,7 % ; IC à 95 % : 47,0 % ; 74,4 %). La réponse des critères de la réponse complète de la MAT a augmenté à 48 patients (85,7 % ; IC à 95 % : 75,7 % ; 95,8 %) pour la normalisation des plaquettes, 47 patients (83,9 % ; IC à 95 % : 73,4 % ; 94,4 %) pour la normalisation de la LDH et 35 patients (62,5 % ; IC à 95 % : 48,9 % ; 76,1 %) pour l'amélioration de la fonction rénale.
Une réponse complète de la MAT a été atteinte après une période médiane de 86 jours (7 à 169 jours). Une augmentation du nombre de plaquettes moyen a été observée rapidement après le début du traitement par le ravulizumab, ce nombre passant de 118,52 × 109/L lors de l'inclusion à 240,34 × 109/L au jour 8 et restant au-dessus de 227 × 109/L lors de toutes les visites ultérieures de la période d'évaluation initiale (26 semaines). De même, le taux moyen de LDH a diminué par rapport à la valeur à l'inclusion au cours des 2 premiers mois de traitement et s'est maintenu pendant toute la période d'évaluation initiale (26 semaines).
Chez les patients présentant une IRC de stade 5, 67,6 % (23/34) ont présenté une amélioration d'au moins un stade de l'IRC. Le stade de l'insuffisance rénale chronique a continué à s'améliorer chez de nombreux patients (19/30) après qu'ils aient obtenu une réponse complète de la MAT pendant la période d'évaluation initiale de 26 semaines. Dix-sept des 29 patients qui avaient besoin de séances de dialyse à l'inclusion dans l'étude étaient en mesure d'arrêter les séances au terme du suivi disponible, alors que 6 des 27 patients qui n'avaient pas besoin de séances de dialyse à l'inclusion étaient sous dialyse lors du dernier suivi disponible. Le tableau 14 résume les critères d'efficacité secondaires de l'étude ALXN1210-aHUS-311.
Tableau 14 : Critères d'efficacité secondaires de l'étude ALXN1210-aHUS-311
| Paramètres |
Étude ALXN1210-aHUS-311 (N = 56) |
|
|
Paramètres
hématologiques relatifs à l a MAT, jour 183 Plaquettes (109/L), sang Moyenne (ET) Médiane LDH (U/L), sérum Moyenne (ET) Médiane |
Valeur observée (n = 48) 237,96 (73,528) 232,00 194,46 (58,099) 176,50 |
Variation par rapport à l'inclusion (n = 48) 114,79 (105,568) 125,00 -519,83 (572,467) -310,75 |
|
Augmentation
de l'hémoglobine ≥ 20 g/L par rapport à l'inclusion avec une confirmation du résultat pendant la période d'évaluation initiale m/n Proportion (IC à 95 %)** |
40/56 0,714 (0,587 ; 0,842) |
|
|
Modification
du stade de l'IRC par rapport à l'inclusion, jour 183 Améliorationa m/n Proportion (IC à 95 %)* Détériorationb m/n Proportion (IC à 95 %)* |
32/47 0,681 (0,529 ; 0,809) 2/13 0,154 (0,019 ; 0,454) |
|
|
DFGe (mL/min/1,73 m2),
jour 183 Moyenne (ET) Médiane |
Valeur observée (n = 48) 51,83 (39,162) 40,00 |
Variation par rapport à l'inclusion (n = 47) 34,80 (35,454) 29,00 |
Note
: n : nombre de patients avec des données disponibles pour une évaluation
donnée lors de la visite du jour 183 ; m : nombre de patients répondant à un
critère donné. Le stade de l'insuffisance rénale chronique (IRC) est déterminé
d'après la classification en stades de l'insuffisance rénale chronique proposée
par la National Kidney Foundation. Le stade 5
correspond au stade terminal et le stade 1 à la forme la
moins avancée de la maladie. Le stade à l'inclusion est déterminé à partir du
dernier DFGe disponible avant le début du traitement.
Amélioration/détérioration : par rapport au stade de l'IRC à l'inclusion. * Les intervalles de confiance à 95 % (IC à 95 %) sont basés sur les limites de confiance exactes déterminées à l'aide de la méthode de Clopper Pearson. a Exclut les patients présentant une IRC de stade 1 à l'inclusion, car aucune amélioration n'est possible. b Exclut les patients présentant une IRC de stade 5 à l'inclusion, car aucune détérioration n'est possible. Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ;
MAT = microangiopathie thrombotique.
Amélioration/détérioration : par rapport au stade de l'IRC à l'inclusion. * Les intervalles de confiance à 95 % (IC à 95 %) sont basés sur les limites de confiance exactes déterminées à l'aide de la méthode de Clopper Pearson. a Exclut les patients présentant une IRC de stade 1 à l'inclusion, car aucune amélioration n'est possible. b Exclut les patients présentant une IRC de stade 5 à l'inclusion, car aucune détérioration n'est possible. Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ;
MAT = microangiopathie thrombotique.
Myasthénie acquise généralisée (MAg)
Étude chez des patients adultes atteints de MAg
L'efficacité et la sécurité du ravulizumab chez les patients adultes atteints de MAg ont été évaluées dans une étude de phase III multicentrique, randomisée en double aveugle, contrôlée contre placebo (étude ALXN1210-MG-306). Les patients participant à cette étude pouvaient ensuite entrer dans une période d'extension en ouvert au cours de laquelle ils ont tous reçu le ravulizumab.
Des patients atteints de MAg (diagnostiqués depuis au moins 6 mois) ayant un test sérologique positif pour les anticorps anti-récepteurs de l'acétylcholine (aRAch), présentant une myasthénie de classes II à IV de la classification clinique de la MGFA (Myasthenia Gravis Foundation of America) et des symptômes persistants, objectivés par un score total ≥ 6 sur l'échelle MG-ADL (Myasthenia Gravis Activities of Daily Living), ont été randomisés pour recevoir le ravulizumab (N = 86) ou le placebo (N = 89). Les patients sous traitements immunosuppresseurs (corticoïdes, azathioprine, cyclophosphamide, ciclosporine, méthotrexate, mycophénolatemofétil ou tacrolimus) pouvaient poursuivre ce traitement pendant toute la durée de l'étude. De plus, un traitement de secours (comprenant corticoïdes à dose élevée, EP/PP ou IgIV) était autorisé si un patient présentait une détérioration clinique, telle que définie par le protocole de l'étude.
Au total, 162 patients (92,6 %) ont terminé la période randomisée contrôlée de 26 semaines de l'étude ALXN1210-MG-306. Les caractéristiques initiales des patients sont présentées dans le tableau 15. La majorité des patients (97 %) inclus dans l'étude avaient reçu au moins un traitement immunomodulateur incluant médicaments immunosuppresseurs, EP/PP ou IgIV au cours des deux années précédant l'inclusion.
Tableau
15 : Caractéristiques cliniques initiales dans l'étude ALXN1210-MG-306
| Paramètre | Statistique |
Placebo (N = 89) |
Ravulizumab (N = 86) |
|
Sexe
Masculin Féminin |
n (%) |
44 (49,4) 45 (50,6) |
42 (48,8) 44 (51,2) |
|
Âge
lors de la première administration du médicament expérimental (ans) |
Moyenne (ET) (min, max) |
53,3 (16,05) (20, 82) |
58,0 (13,82) (19, 79) |
|
Patients
âgés de 65 ans et plus lors de l'inclusion dans l'étude |
n (%) | 24 (27,0) | 30 (34,9) |
|
Ancienneté
de la MAg depuis le diagnostic (ans) |
Moyenne (ET) (min, max) Médiane |
10,0 (8,90) (0,5, 36,1) 7,6 |
9,8 (9,68) (0,5, 39,5) 5,7 |
| Score MG-ADL lors de l'inclusion |
Moyenne (ET) (min, max) Médiane |
8,9 (2,30) (6,0, 15,0) 9,0 |
9,1 (2,62) (6,0, 24,0) 9,0 |
| Score QMG lors de l'inclusion |
Moyenne (ET) (min, max) Médiane |
14,5 (5,26) (2,0, 27,0) 14,0 |
14,8 (5,21) (6,0, 39,0) 15,0 |
|
Classification
MGFA lors de l'inclusion
Classe II (faiblesse légère) Classe III (faiblesse modérée) Classe IV (faiblesse sévère) |
n (%) |
39 (44) 45 (51) 5 (6) |
39 (45) 41 (48) 6 (7) |
|
Antécédents
d'intubation depuis le diagnostic (classe V de la MGFA) |
n (%) | 9 (10,1) | 8 (9,3) |
|
Nombre
de patients ayant présenté des poussées de la MA depuis le diagnostica |
n (%) | 17 (19,1) | 21 (24,4) |
|
Nombre
de traitements immunosuppresseurs stablesb lors de l'inclusion dans l'étude 0 1 ≥ 2 |
n (%) |
8 (9,0) 34 (38,2) 47 (52,8) |
10 (11,6) 40 (46,5) 36 (41,9) |
b Les traitements immunosuppresseurs comprennent : corticoïdes, azathioprine, cyclophosphamide, ciclosporine, méthotrexate, mycophénolatemofétil ou tacrolimus.
Abréviations : max = maximum ; min =
minimum ; MA = myasthénie acquise ; MGADL = Myasthenia
Gravis Activities of Daily Living - échelle
d'évaluation du retentissement de la myasthénie acquise sur les activités de la
vie quotidienne ; MGFA = Myasthenia Gravis Foundation of America ; QMG = Quantitative
Myasthenia Gravis ; ET = écart type.
Le critère d'évaluation principal était la variation du score MG-ADL total à la semaine 26 par rapport à l'inclusion.
Les critères d'évaluation secondaires, portant également sur les variations par rapport à l'inclusion à la semaine 26, étaient la variation du score total de l'échelle QMG (Quantitative Myasthenia Gravis), le pourcentage de patients présentant des améliorations d'au moins 5 points et 3 points respectivement des scores QMG et MG-ADL totaux, ainsi que les variations des scores d'évaluation de la qualité de vie.
Par rapport au placebo, une variation statistiquement significative du score MG-ADL total a été observée avec le ravulizumab. Les résultats des critères d'évaluation principal et secondaires sont présentés dans le tableau 16.
Tableau
16 : Analyses des critères d'évaluation de l'efficacité principal et
secondaires
|
Critères
d'évaluation de l'efficacité à la semaine 26 |
Placebo (N = 89) Moyenne des MC (ETM) |
Ravulizumab
(N = 86) Moyenne des MC (ETM) |
Statistique pour la comparaison |
Effet du traitement (IC à 95 %) |
Valeur p (selon un modèle à effets mixtes pour mesures répétées) |
| MG-ADL | -1,4 (0,37) | -3,1 (0,38) |
Différence de la variation
par rapport au score initial |
-1,6 (-2,6 ; -0,7) | 0,0009 |
| QMG | -0,8 (0,45) | -2,8 (0,46) |
Différence de la variation
par rapport au score initial |
-2,0 (-3,2 ; -0,8) | 0,0009 |
| MG-QoL15r | -1,6 (0,70) | -3,3 (0,71) |
Différence de la variation
par rapport au score initial |
-1,7 (-3,4 ; 0,1) | 0,0636 |
| Neuro-QoL-fatigue | -4,8 (1,87) | -7,0 (1,92) |
Différence
de la variation par rapport au score initial |
-2,2 (-6,9 ; 2,6) | 0,3734a |
a Il
n'a pas été effectué de tests formels de la significativité statistique pour le
critère d'évaluation ; une valeur p nominale était présentée.
Abréviations : IC = intervalle de confiance ; MC = moindres carrés ; MG-ADL = Myasthenia Gravis Activities of Daily Living ; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item scale - échelle d'évaluation en 15 items de la qualité de vie dans la myasthénie acquise révisée ; Neuro-QoL-fatigue = Neurological Quality of Life Fatigue - échelle d'évaluation de la qualité de vie dans les maladies neurologiques ; QMG = Quantitative Myasthenia Gravis ; ETM = erreur type de la moyenne.
Abréviations : IC = intervalle de confiance ; MC = moindres carrés ; MG-ADL = Myasthenia Gravis Activities of Daily Living ; MG-QoL15r = Revised Myasthenia Gravis Quality of Life 15-item scale - échelle d'évaluation en 15 items de la qualité de vie dans la myasthénie acquise révisée ; Neuro-QoL-fatigue = Neurological Quality of Life Fatigue - échelle d'évaluation de la qualité de vie dans les maladies neurologiques ; QMG = Quantitative Myasthenia Gravis ; ETM = erreur type de la moyenne.
Dans l'étude ALXN1210-MG-306, un répondeur au traitement pour le score MG-ADL total était défini comme un patient présentant une amélioration d'au moins 3 points. Le pourcentage de répondeurs au traitement à la semaine 26 était de 56,7 % dans le groupe ravulizumab contre 34,1 % dans le groupe placebo (valeur p nominale = 0,0049). Un répondeur au traitement pour le score QMG total était défini comme un patient présentant une amélioration d'au moins 5 points. Le pourcentage de répondeurs au traitement à la semaine 26 était de 30,0 % dans le groupe ravulizumab contre 11,3 % dans le groupe placebo (p = 0,0052).
Le tableau 17 présente une vue d'ensemble des patients ayant présenté une détérioration clinique et des patients ayant eu besoin d'un traitement de secours pendant la période randomisée contrôlée de 26 semaines.
Tableau 17 : Détérioration clinique et traitement de secours
| Paramètre | Statistique |
Placebo
(N = 89) |
Ravulizumab (N = 86) |
|
Nombre
total de patients ayant présenté une détérioration clinique |
n (%) | 15 (16,9) | 8 (9,3) |
|
Nombre
total de patients ayant eu besoin d'un traitement de secoursa |
n (%) | 14 (15,7) | 8 (9,3) |
a Les
traitements de secours comprenaient : corticoïdes à dose élevée, échange
plasmatique/plasmaphérèse ou immunoglobulines intraveineuses.
Au moment de l'analyse, 150 des 158 patients qui étaient entrés dans la période d'extension en ouvert étaient toujours dans l'étude.
Chez les patients qui avaient reçu initialement Ultomiris pendant la période randomisée contrôlée et qui ont poursuivi le traitement pendant les 34 premières semaines de la période d'extension en ouvert, l'effet du traitement a été maintenu (figure 3). Chez les patients qui avaient reçu initialement le placebo pendant la période randomisée contrôlée de 26 semaines et qui ont commencé le traitement par Ultomiris pendant la période d'extension en ouvert, une réponse rapide et maintenue au traitement a été observée (figure 3).
Figure 3 : Variation du score MG-ADL total (A) et du score QMG total (B) jusqu'à la semaine 60 par rapport à l'inclusion dans la période randomisée contrôlée (moyenne et IC à 95 %)
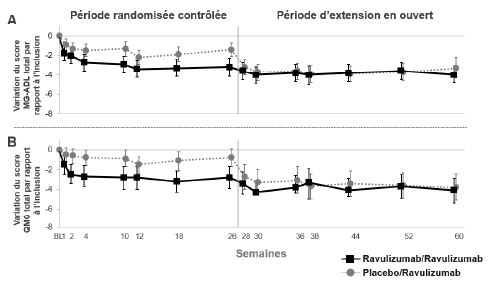
Abréviations : IC = intervalle de confiance ; MG-ADL = Myasthenia Gravis Activities of Daily Living ; QMG = Quantitative Myasthenia Gravis.
Dans la période d'extension en ouvert de l'étude, les cliniciens avaient la possibilité d'ajuster les traitements immunosuppresseurs. Chez les patients suivis pendant 34 semaines dans la période d'extension en ouvert, la dose quotidienne de corticoïdes a été diminuée chez 28,0 % des patients et la corticothérapie a été arrêtée chez 6,2 %. La raison la plus fréquente de modification de la corticothérapie était l'amélioration des symptômes de la MA pendant le traitement par le ravulizumab.
Maladie du spectre de la neuromyélite optique (NMOSD)
Étude chez des patients adultes atteints de NMOSD
L'efficacité et la sécurité du ravulizumab chez les patients adultes atteints de NMOSD présentant des anticorps anti-AQP4 ont été évaluées dans une étude internationale en ouvert (ALXN1210-NMO-307).
L’étude ALX1210-NMO-307 a été menée chez 58 patients adultes atteints de NMOSD ayant une sérologie positive pour les anticorps anti-AQP4, qui avaient présenté au moins une poussée au cours des 12 mois précédant la période de sélection et qui avaient un score ≤ 7 sur l’échelle EDSS (Expanded Disability Status Scale). Un traitement antérieur par des immunosuppresseurs n’était pas une condition requise pour l’inclusion et 51,7 % des patients ont reçu le ravulizumab en monothérapie. Les patients recevant certains traitements immunosuppresseurs (par exemple : corticoïdes, azathioprine, mycophénolate mofétil, tacrolimus) pouvaient poursuivre le traitement en association avec le ravulizumab, la dose devant être stable jusqu’à ce qu’ils aient atteint la semaine 106 de l’étude. De plus, un traitement aigu d’une poussée (incluant corticoïdes à dose élevée, EP/PP et IgIV) était autorisé si un patient présentait une poussée pendant l’étude.
L'âge moyen des patients inclus dans l'étude était de 47,4 ans (intervalle : 18 à 74 ans) et la majorité des patients étaient des femmes (90 %). L'âge médian lors des premières manifestations cliniques de la NMOSD était de 42,5 ans (intervalle : 16 à 73 ans). Les caractéristiques cliniques initiales sont présentées dans le tableau 18.
Tableau
18 : Antécédents médicaux liés à la maladie et caractéristiques initiales des
patients dans l'étude ALXN1210-NMO-307
| Paramètre | Statistique |
ALXN1210-NMO-307
Ravulizumab (N = 58) |
|
Délai
entre les premières manifestations cliniques de la NMOSD et la première dose de médicament expérimental (années) |
Moyenne (ET) | 5,2 (6,38) |
| Médiane | 2,0 | |
| Min, max | 0,19, 24,49 | |
|
TAP
historique au cours des 24 mois précédant la sélection |
Moyenne (ET) | 1,87 (1,59) |
| Médiane | 1,44 | |
| Min, max | 0,5, 6,9 | |
| Score HAI à l'inclusion | Moyenne (ET) | 1,2 (1,42) |
| Médiane | 1,0 | |
| Min, max | 0, 7 | |
| Score EDSS à l'inclusion | Moyenne (ET) | 3,30 (1,58) |
| Médiane | 3,25 | |
| Min, max | 0,0, 7,0 | |
| Tout traitement antérieur par rituximab | n (%) | 21 (36,2) |
|
Nombre
de patients ne recevant que des corticoïdes à dose stable lors de l'inclusion dans l'étude |
n (%) | 12 (20,7) |
|
Nombre
de patients ne recevant aucun TIS lors de l'inclusion dans l'étude |
n (%) | 30 (51,7) |
Abréviations
: TAP = taux annualisé de poussées ; EDSS = Expanded
Disability Status Scale (échelle d'évaluation du handicap) ; HAI = Hauser
Ambulation Index (échelle d'évaluation quantitative de la marche) ; TIS =
traitement immunosuppresseur ; max = maximum ; min = minimum ; NMOSD = maladie
du spectre de la neuromyélite optique ; ET =
écart-type.
Le critère d'évaluation principal de l'étude ALXN1210-NMO-307 était le délai jusqu'à la première poussée survenant pendant l'étude, confirmée par un comité d'évaluation indépendant. Pendant la période de traitement principale, il n'a pas été observé de poussée confirmée chez les patients traités par le ravulizumab. Pendant le suivi d'une durée médiane de 90,93 semaines, aucun des patients traités par le ravulizumab n'a présenté de poussée. Les résultats du critère d'évaluation principal, l'absence de poussées, étaient concordants chez les patients traités par le ravulizumab avec ou sans traitement immunosuppresseur concomitant.
Le ravulizumab n'a pas été étudié dans le traitement aigu des poussées chez les patients atteints de NMOSD.
Population pédiatrique
Hémoglobinurie paroxystique nocturne (HPN)
Étude chez des patients pédiatriques atteints d'HPN (ALXN1210-PNH-304)
L'étude
pédiatrique (ALXN1210-PNH-304) est une étude de phase III multicentrique en
ouvert menée chez des enfants et adolescents atteints d'HPN naïfs d'inhibiteur
du complément ou préalablement traités par l'eculizumab.
Selon les résultats intermédiaires, 13 patients pédiatriques atteints d'HPN au total
ont terminé le traitement par le ravulizumab pendant
la période d'évaluation principale (26 semaines) de l'étude ALXN1210-PNH-304.
Cinq des 13 patients n'avaient jamais reçu d'inhibiteur du complément et 8
patients étaient traités par l'eculizumab avant
l'inclusion dans l'étude.
La majorité des patients étaient âgés de 12 ans à 17 ans au moment de la première perfusion (moyenne : 14,4 ans), deux patients étant âgés de moins de 12 ans (11 ans et 9 ans). Huit des 13 patients étaient de sexe féminin. Le poids moyen à l'inclusion était de 56 kg (37 kg à 72 kg). Le tableau 19 présente les antécédents médicaux liés à la maladie et les caractéristiques initiales des patients pédiatriques inclus dans l'étude ALXN1210-PNH-304.
Tableau 19 : Antécédents médicaux liés à la maladie et caractéristiques initiales
(population complète d'analyse)
| Paramètre |
Patients naïfs d'inhibiteur du complément (N = 5) |
Patients préalablement
traités par l'eculizumab (N = 8) |
|
Taille
totale du clone HPN érythrocytaire (%) Médiane (min, max) |
(N = 4) 40,05 (6,9 ; 68,1) |
N = 6 71,15 (21,2 ; 85,4) |
|
Taille
totale du clone HPN granulocytaire (%) Médiane (min, max) |
78,30 (36,8 ; 99,0) |
91,60 (20,3 ; 97,6) |
|
Nombre
de patients ayant reçu des transfusions de concentré érythrocytaire/sang total au cours des 12 mois précédant la première dose, n (%) |
2 (40,0) | 2 (25,0) |
|
Nombre
de transfusions de concentré érythrocytaire/sang total au cours des 12 mois précédant la première dose Total Médiane (min, max) |
10 5,0 (4, 6) |
2 1,0 (1, 1) |
|
Unités
de concentré érythrocytaire/sang total transfusées au cours des 12 mois précédant la première dose Total Médiane (min, max) |
14 7,0 (3, 11) |
2 2,0 (2, 2) |
|
Patients
présentant des pathologies liées à l'HPN avant le consentement éclairé, n (%) Anémie Hématurie ou hémoglobinurie Anémie aplasique Insuffisance rénale Autrea |
5 (100) 2 (40,0) 2 (40,0) 3 (60,0) 2 (40,0) 0 |
8 (100) 5 (62,5) 5 (62,5) 1 (12,5) 2 (25,0) 1 (12,5) |
|
Taux
de LDH avant traitement (U/L) Médiane (min, max) |
588,50 (444 ; 2269,7) |
251,50 (140,5 ; 487) |
a Les
autres pathologies liées à l'HPN étaient rapportées avec les termes « infarctus
rénal et splénique » et « lésions multiples évoquant un processus embolique ».
Note : les pourcentages sont basés sur le nombre total de patients dans chaque cohorte.
Abréviations : LDH = lactate déshydrogénase ; max = maximum ; min = minimum ; HPN= hémoglobinurie paroxystique nocturne.
Note : les pourcentages sont basés sur le nombre total de patients dans chaque cohorte.
Abréviations : LDH = lactate déshydrogénase ; max = maximum ; min = minimum ; HPN= hémoglobinurie paroxystique nocturne.
Les patients ont reçu une dose de charge de ravulizumab déterminée en fonction du poids le jour 1, suivie d'un traitement d'entretien administré le jour 15, puis toutes les 8 semaines (q8s) chez les patients pesant ≥ 20 kg ou toutes les 4 semaines (q4s) chez les patients pesant < 20 kg. Chez les patients sous traitement par l'eculizumab au moment de l'inclusion dans l'étude, le jour 1 du traitement de l'étude était planifié deux semaines après l'administration de la dernière dose d'eculizumab.
Le
ravulizumab administré selon un schéma posologique en
fonction du poids a conféré une inhibition immédiate, complète et maintenue de
la voie terminale du complément pendant toute la période d'évaluation
principale de 26 semaines, que les patients aient été ou non préalablement
traités par l'eculizumab. Après l'instauration du
traitement par le ravulizumab, les concentrations
sériques du ravulizumab à l'état d'équilibre ont été
atteintes immédiatement après la première dose et se sont maintenues pendant
toute la période d'évaluation principale de 26 semaines dans les deux cohortes.
Il n'a pas été observé d'épisodes hémolytiques dans l'étude et aucun patient n'a
eu un taux de C5 libre supérieur à 0,5 µg/mL après
l'inclusion. Pendant la période d'évaluation principale de 26 semaines, la
variation relative moyenne du taux de LDH par rapport à l'inclusion était de
-47,91 % le jour 183 dans la cohorte de patients naïfs d'inhibiteur du
complément et le taux est resté stable dans la cohorte de patients
préalablement traités par l'eculizumab. Soixante pour
cent des patients naïfs d'inhibiteur du complément (3/5) et 75 % des patients
préalablement traités par l'eculizumab (6/8)
respectivement avaient obtenu une stabilisation de l'hémoglobine à la semaine
26. L'absence de recours à la transfusion a été rapportée chez 84,6 % des
patients (11/13) pendant la période d'évaluation principale de 26 semaines.
Ces résultats intermédiaires des critères d'efficacité sont présentés dans le
tableau 20 ci-dessous.
Tableau
20 : Résultats des critères d'efficacité dans l'étude
pédiatrique chez des patients atteints d'HPN (ALXN1210-PNH-304) - période
d'évaluation principale de 26 semaines
| Critère |
Ravulizumab (naïfs de traitement, N = 5) |
Ravulizumab
(relais de l'eculizumab, N = 8) |
|
LDH
- variation relative par rapport à l'inclusion Moyenne (ET) |
-47,91 (52,716) |
4,65 (44,702) |
|
Absence
de recours à la transfusion Pourcentage (IC à 95 %) |
60,0 (14,66 ; 94,73) |
100,0 (63,06 ; 100,00) |
|
Stabilisation
de l'hémoglobine Pourcentage (IC à 95 %) |
60,0 (14,66 ; 94,73) |
75 (34,91 ; 96,81) |
| Épisodes hémolytiques (%) | 0 | 0 |
Abréviation
: LDH = lactate déshydrogénase.
Les résultats d’efficacité à long terme jusqu’à la fin de l’étude, sur une durée médiane de traitement de 915 jours, ont montré une réponse au traitement maintenue chez les patients pédiatriques atteints d’HPN.
Selon les données de ces résultats intermédiaires, l'efficacité du ravulizumab chez les patients pédiatriques atteints d'HPN semble être similaire à celle observée chez les patients adultes.
Syndrome hémolytique et urémique atypique (SHUa)
L'utilisation d'Ultomiris pour le traitement du SHUa chez les patients pédiatriques est étayée par des
données issues d'une étude clinique pédiatrique (au total, 31 patients atteints
de SHUa documenté ont été inclus. Vingt-huit patients
âgés de 10 mois à 17 ans ont été inclus dans la population complète d'analyse).
Étude chez les patients pédiatriques atteints de SHUa (ALXN1210-aHUS-312)
L'étude
pédiatrique est une étude en cours, de phase III, multicentrique, à bras
unique, d'une durée de 26 semaines menée chez des patients pédiatriques.
Au total, 21 patients naïfs de traitement par l'eculizumab
avec un diagnostic documenté de SHUa et des signes de
MAT ont été inclus ; parmi ces patients, 18 ont été inclus dans la population
complète d'analyse. Selon les critères d'inclusion, les patients présentant une
MAT due à un PTT ou à un SHU à STEC étaient exclus. Deux patients ont reçu une
seule dose et un patient a reçu 2 doses ; ils ont ensuite arrêté le traitement
et ont été exclus de la population d'analyse complète car le SHUa n'a pas été confirmé. Le poids moyen à l'inclusion
était de 22,2 kg ; la majorité des patients relevaient de la catégorie de poids
à l'inclusion de ≥ 10 à < 20 kg. La majorité des patients (72,2 %)
présentaient des signes (aux niveaux cardiovasculaire, pulmonaire, du système
nerveux central, gastro-intestinal, cutané et musculo-squelettique) ou des
symptômes extrarénaux de SHUa lors de l'inclusion. À
l'inclusion, 33,3 % (n = 6) des patients présentaient une IRC de stade 5.
Au total, 10 patients chez lesquels un relais de l'eculizumab par le ravulizumab a été effectué et qui présentaient un diagnostic documenté de SHUa et des signes de MAT ont été inclus. Les patients devaient présenter une réponse clinique à l'eculizumab avant l'inclusion (c'est-à-dire, taux de LDH < 1,5 × LSN et nombre de plaquettes ≥ 150 000/µL et DFGe > 30 mL/min/1,73 m2). Par conséquent, il n'existe aucune information sur l'utilisation du ravulizumab chez les patients réfractaires à l'eculizumab.
Le tableau 21 présente les caractéristiques initiales des patients pédiatriques inclus dans l'étude ALXN1210-aHUS-312.
Tableau
21 : Caractéristiques démographiques et initiales dans l'étude
ALXN1210-aHUS-312
| Paramètre | Statistiques |
Ravulizumab
(naïfs, N = 18) |
Ravulizumab
(relais, N = 10) |
|
Tranche
d'âge (ans) lors de la première perfusion Naissance à < 2 ans 2 ans à < 6 ans 6 ans à < 12 ans 12 ans à < 18 ans |
n (%) |
2 (11,1) 9 (50,0) 5 (27,8) 2 (11,1) |
1 (10,0) 1 (10,0) 1 (10,0) 7 (70,0) |
|
Sexe
Masculin |
n (%) |
8 (44,4) |
9 (90,0) |
|
Origine
ethniquea Amérindienne ou autochtone de l'Alaska Asiatique Noire ou afro-américaine Caucasienne Inconnue |
n (%) |
1 (5,6) 5 (27,8) 3 (16,7) 9 (50,0) 1 (5,6) |
0 (0,0) 4 (40,0) 1 (10,0) 5 (50,0) 0 (0,0) |
| Antécédents de greffe | n (%) | 1 (5,6) | 1 (10,0) |
| Plaquettes (109/L), sang | Médiane (min, max) | 51,25 (14, 125) | 281,75 (207,415,5) |
| Hémoglobine (g/L) | Médiane (min, max) | 74,25 (32, 106) | 132,0 (114,5,148) |
| LDH (U/L) | Médiane (min,max) | 1 963,0 (772,4 985) | 206,5 (138,5,356) |
| DFGe (mL/min/1,73 m2) | Médiane (min, max) | 22,0 (10, 84) | 99,75 (54,136,5) |
| Dialyse nécessaire à l'inclusion | n (%) | 6 (33,3) | 0 (0,0) |
Note
: les pourcentages sont basés sur le nombre total de patients.
a Les patients peuvent présenter plusieurs origines ethniques.
Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ; max = maximum ; min = minimum.
a Les patients peuvent présenter plusieurs origines ethniques.
Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ; max = maximum ; min = minimum.
Le critère d'évaluation principal était la réponse complète de la MAT pendant la période d'évaluation initiale de 26 semaines, attestée par une normalisation des paramètres hématologiques (plaquettes ≥ 150 × 109/L et LDH ≤ 246 U/L) et une amélioration ≥ 25 % de la créatininémie par rapport à l'inclusion. Les patients devaient satisfaire à tous les critères de réponse complète de la MAT lors de 2 évaluations distinctes réalisées à intervalle d'au moins 4 semaines (28 jours) et lors de toute mesure intermédiaire.
Une réponse complète de la MAT a été observée chez 14 des 18 patients naïfs de traitement (77,8 %) pendant la période d'évaluation initiale de 26 semaines, comme indiqué dans le tableau 22.
Tableau
22 : Analyse de la réponse complète de la MAT et des composantes de la réponse
complète de la MAT pendant la période d'évaluation initiale de 26 semaines
(ALXN1210-aHUS-312)
| Total | Répondeurs | ||
| n | Proportion (IC à 95 %)a | ||
| Réponse complète de la MAT | 18 | 14 | 0,778 (0,524 ; 0,936) |
|
Composante
de la réponse complète de la MAT Normalisation des plaquettes Normalisation de la LDH Amélioration ≥ 25 % de la créatininémie par rapport à l'inclusion |
18 18 18 |
17 16 15 |
0,944 (0,727 ; 0,999) 0,889 (0,653 ; 0,986) 0,833 (0,586 ; 0,964) |
| Normalisation hématologique | 18 | 16 | 0,889 (0,653 ; 0,986) |
Note
: un patient est sorti de l'étude après avoir reçu deux doses de ravulizumab.
a Les IC à 95 % de la proportion étaient basés sur la méthode d'approximation asymptotique gaussienne avec une correction de continuité.
Abréviations : IC = intervalle de confiance ; LDH = lactate déshydrogénase ; MAT = microangiopathie thrombotique.
a Les IC à 95 % de la proportion étaient basés sur la méthode d'approximation asymptotique gaussienne avec une correction de continuité.
Abréviations : IC = intervalle de confiance ; LDH = lactate déshydrogénase ; MAT = microangiopathie thrombotique.
Pendant la période d'évaluation initiale, une réponse complète de la MAT a été atteinte après une période médiane de 30 jours (15 à 97 jours). Tous les patients présentant une réponse complète de la MAT l'ont conservée pendant toute la période d'évaluation initiale, avec une amélioration continue de la fonction rénale. Une augmentation du nombre de plaquettes moyen a été observée rapidement après le début du traitement par le ravulizumab, ce nombre passant de 60,50 × 109/L lors de l'inclusion à 296,67 × 109/L au jour 8 et restant au-dessus de 296 × 109/L lors de toutes les visites ultérieures au cours de la période d'évaluation initiale (26 semaines).
Trois patients supplémentaires ont présenté une réponse complète de la MAT, qui a été confirmée après la période d'évaluation initiale de 26 semaines (avec une réponse complète de la MAT survenant aux jours 291, 297 et 353) ; ainsi, 17 patients sur 18 (94,4 %) (IC à 95 % : 72,7 % ; 99,9 %) ont présenté une réponse complète de la MAT. La réponse des composantes individuelles a augmenté et est passée à 17 patients sur 18 (94,4 % ; IC à 95 % : 72,7 % ; 99,9 %) pour la normalisation du nombre de plaquettes, 17 patients sur 18 (94,4 % ; IC à 95 % : 72,7 % ; 99,9 %) pour la normalisation de la LDH et 17 patients sur 18 (94,4 % ; IC à 95 % : 72,7 % ; 99,9 %) pour l'amélioration de la fonction rénale.
Les 6 patients qui avaient besoin de séances de dialyse lors de leur inclusion dans l'étude étaient en mesure d'arrêter les séances ; 5 d'entre eux les avaient déjà arrêtées au jour 43. Aucun patient n'a commencé de dialyse pendant l'étude. La majorité de la population de patients (15/17) présentait une amélioration d'au moins 1 stade de l'IRC au jour 183 ; 14 patients présentaient une amélioration d'au moins deux stades. Le tableau 23 résume les résultats des critères d'efficacité secondaires de l'étude ALXN1210-aHUS-312.
Tableau 23 : Critère d'efficacité secondaire de l'étude ALXN1210-aHUS-312.
| Paramètres | Étude ALXN1210-aHUS-312 (N = 18) | |
|
Paramètres
hématologiques relatifs à la MAT, jour 183 Plaquettes (109/L), sang Moyenne (ET) Médiane LDH (U/L), sérum Moyenne (ET) Médiane |
Valeur observée (n = 17) 304,94 (75,711) 318,00 262,41 (59,995) 247,00 |
Variation par rapport à
l'inclusion (n = 17) 245,59 (91,827) 247,00 -2 044,13 (1 328,059) -1 851,50 |
|
Augmentation
de l'hémoglobine ≥ 20 g/L par rapport à l'inclusion avec une confirmation du résultat pendant la période d'évaluation initiale m/N Proportion (IC à 95 %)* |
16/18 0,889 (0,653 ; 0,986) |
|
|
Modification
du stade de l'IRC par rapport à l'inclusion, jour 183 Améliorationa m/n Proportion (IC à 95 %)* Détériorationb m/n Proportion (IC à 95 %)* |
15/17 0,882 (0,636 ; 0,985) 0/11 0,000 (0,000 ; 0,285) |
|
|
DFGe (mL/min/1,73 m2),
jour 183 Moyenne (ET) Médiane |
Valeur observée (n = 17) 108,5 (56,87) 108,0 |
Variation par rapport à
l'inclusion (n = 17) 85,4 (54,33) 80,0 |
Note
: n : nombre de patients avec des données disponibles pour une évaluation
donnée lors de la visite du jour 183 ; m : nombre de patients répondant à un
critère donné. Le stade de l'insuffisance rénale chronique (IRC) est déterminé
d'après la classification en stades de l'insuffisance rénale chronique proposée
par la National Kidney Foundation.
Le stade 1 correspond au stade le moins avancé de la maladie et le stade 5 au
stade terminal. Le stade à l'inclusion est déterminé à partir du dernier DFGe disponible avant le début du traitement.
Amélioration/détérioration : par rapport au stade de l'IRC à l'inclusion.
* Les intervalles de confiance à 95 % (IC à 95 %) sont basés sur les limites de confiance exactes déterminées à l'aide de la méthode de Clopper Pearson.
a La catégorie « amélioration » exclut les patients présentant une IRC de stade 1 à l'inclusion, car aucune amélioration n'est possible.
b La catégorie « détérioration » exclut les patients présentant une IRC de stade 5 à l'inclusion, car aucune détérioration n'est possible.
Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ; MAT = microangiopathie thrombotique.
Amélioration/détérioration : par rapport au stade de l'IRC à l'inclusion.
* Les intervalles de confiance à 95 % (IC à 95 %) sont basés sur les limites de confiance exactes déterminées à l'aide de la méthode de Clopper Pearson.
a La catégorie « amélioration » exclut les patients présentant une IRC de stade 1 à l'inclusion, car aucune amélioration n'est possible.
b La catégorie « détérioration » exclut les patients présentant une IRC de stade 5 à l'inclusion, car aucune détérioration n'est possible.
Abréviations : DFGe = débit de filtration glomérulaire estimé ; LDH = lactate déshydrogénase ; MAT = microangiopathie thrombotique.
Chez les patients préalablement traités par l'eculizumab, un relais de l'eculizumab par le ravulizumab a permis de maintenir le contrôle de la maladie, comme le montrent les paramètres hématologiques et rénaux stables, sans incidence notable sur la sécurité.
L'efficacité du ravulizumab dans le traitement du SHUa semble similaire chez les patients pédiatriques et les patients adultes.
Myasthénie acquise généralisée (MAg)
L'agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Ultomiris dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement de la myasthénie acquise généralisée (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Maladie du spectre de la neuromyélite optique (NMOSD)
L'agence européenne des médicaments a différé l'obligation de soumettre les résultats d'études réalisées avec Ultomiris dans un ou plusieurs sous-groupes de la population pédiatrique dans le traitement de la NMOSD (voir rubrique Posologie et mode d'administration pour les informations concernant l'usage pédiatrique).
Absorption
La voie d’administration étant une perfusion intraveineuse et la forme pharmaceutique étant une solution, 100 % de la dose de ravulizumab administrée sont considérés comme biodisponibles. Le temps jusqu’à la concentration maximale observée (tmax) est prévu à la fin de la perfusion (FDP) ou peu de temps après la FDP. Les concentrations thérapeutiques du médicament à l’état d’équilibre sont atteintes après la première dose.
Distribution
Le volume central et le volume de distribution moyens (écart-type [ET]) à l'état d'équilibre chez les patients adultes et pédiatriques atteints d'HPN ou de SHUa et chez les patients adultes atteints de MAg ou de NMOSD sont présentés dans le tableau 24.
Biotransformation et élimination
Le ravulizumab étant un anticorps monoclonal de type immunoglobuline gamma (IgG), il devrait être métabolisé de la même manière que toute IgG endogène (dégradé en petits peptides et acides aminés par des voies cataboliques) et être éliminé de la même façon. Le ravulizumab contient uniquement des acides aminés naturellement présents et n'a pas de métabolites actifs connus. Les valeurs moyennes (ET) de la demi-vie d'élimination terminale et de la clairance du ravulizumab chez les patients adultes et pédiatriques atteints d'HPN, chez les patients adultes et pédiatriques atteints de SHUa et chez les patients adultes atteints de MAg ou de NMOSD sont présentées dans le tableau 24.
Tableau
24 : Estimations des paramètres de volume central, de distribution, de
biotransformation et d'élimination après l'administration de ravulizumab
| Patients adultes et pédiatriques atteints d'HPN | Patients adultes et pédiatriques atteints de SHUa | Patients adultes atteints de MAg | Patients adultes atteints deNMOSD | |
|
Volume
central estimé (litres)
Moyenne (ET) |
Patients adultes : 3,44
(0,66)
Patients pédiatriques : 2,87 (0,60) |
Patients adultes : 3,25
(0,61)
Patients pédiatriques : 1,14 (0,51) |
3,42 (0,756) | 2,91 (0,571) |
|
Volume
de distribution à l'état d'équilibre (litres)
Moyenne (ET) |
5,30 (0,9) | 5,22 (1,85) | 5,74 (1,16) | 4,77 (0,819) |
|
Demi-vie
d'élimination terminale (jours) Moyenne (ET) |
49,6 (9,1) | 51,8 (16,2) | 56,6 (8,36) | 64,3 (11,0) |
|
Clairance
(litres/jour) Moyenne (ET) |
0,08 (0,022) | 0,08 (0,04) | 0,08 (0,02) | 0,05 (0,016) |
Abréviations
: SHUa = syndrome hémolytique et urémique atypique ; MAg = myasthénie acquise généralisée ; HPN = hémoglobinurie
paroxystique nocturne ; NMOSD = maladie du spectre de la neuromyélite
optique ; ET = écart-type.
Linéarité/non-linéarité
Dans l'intervalle de doses et de posologies étudiées, la pharmacocinétique (PK) du ravulizumab était proportionnelle à la dose et linéaire dans le temps.
Populations particulières
Poids
Le
poids est une covariable significative chez les
patients atteints d'HPN, de SHUa, de MAg ou de NMOSD, conduisant à des niveaux d'exposition plus
faibles chez les patients ayant un poids plus élevé. La posologie en fonction
du poids est proposée à la rubrique Posologie et mode d'administration,
tableau 1, tableau 3 et tableau 4.
Il
n'a pas été effectué d'étude formelle de l'effet du sexe, de l'origine
ethnique, de l'âge (population gériatrique), de l'insuffisance hépatique ou
rénale sur la pharmacocinétique du ravulizumab.
Toutefois,
l'analyse PK de population n'a pas mis en évidence d'effet du sexe, de l'âge,
de l'origine ethnique et de l'insuffisance hépatique ou rénale sur la PK du ravulizumab chez les volontaires sains et les patients
atteints d'HPN, de SHUa, de MAg
ou de NMOSD étudiés ; par conséquent, aucun ajustement de la dose n'est
considéré comme nécessaire.
La pharmacocinétique du ravulizumab a été étudiée chez des patients atteints de SHUa présentant divers stades d'insuffisance rénale, y compris les patients dialysés. Aucune différence n'a été observée au niveau des paramètres pharmacocinétiques dans ces sous-populations de patients, y compris chez les patients présentant une protéinurie.
Ultomiris n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines.
Il n'a pas été effectué d'études de toxicité du ravulizumab sur la reproduction chez l'animal, mais des études ont été réalisées chez la souris avec un anticorps analogue murin inhibiteur de la voie terminale du complément, le BB5.1. Aucun effet clairement lié au traitement, ni aucun effet indésirable n'a été observé lors des études de toxicité sur la reproduction effectuées chez la souris avec un anticorps analogue murin inhibiteur de la voie terminale du complément. Lors d'une exposition maternelle à l'anticorps au cours de l'organogenèse, deux cas de dysplasie rétinienne et un cas de hernie ombilicale ont été observés parmi les 230 descendants de mères exposées à la dose d'anticorps la plus élevée (environ 4 fois la dose maximale recommandée de ravulizumab chez l'homme, sur la base d'une comparaison des poids) ; en revanche, l'exposition n'a pas augmenté le taux de pertes fœtales, ni la mortalité néonatale.
Il n'a pas été effectué d'études chez l'animal pour évaluer le potentiel génotoxique et cancérogène du ravulizumab.
Les données non cliniques issues des études effectuées avec une molécule murine analogue, le BB5.1, n'ont pas révélé de risque particulier pour l'homme.
Chaque flacon est à usage unique.
Ultomiris 300 mg/3 mL et 1 100 mg/11 mL solutions à diluer pour perfusion
Ce médicament doit être dilué à une concentration finale de 50 mg/mL. Une technique aseptique doit être utilisée.
Préparer Ultomiris solution à diluer pour perfusion en suivant les instructions suivantes :
- Le nombre de flacons à utiliser pour la dilution est déterminé en fonction du poids du patient et de la dose prescrite, voir la rubrique Posologie et mode d'administration.
- Avant dilution, la solution contenue dans les flacons doit être visuellement inspectée pour vérifier l'absence de particules ou de précipité. Ne pas utiliser si des particules ou un précipité sont observés.
- Le volume calculé de médicament est prélevé du nombre de flacons approprié et dilué dans une poche pour perfusion en utilisant comme diluant une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %). Voir ci-dessous les tableaux de référence pour l'administration. Mélanger doucement le produit. Ne pas agiter le produit.
- Après dilution, la concentration finale de la solution à perfuser est de 50 mg/mL.
- La solution préparée doit être administrée immédiatement après la préparation, à moins qu'elle ne soit conservée à une température comprise entre 2 °C et 8 °C. En cas de conservation à une température comprise entre 2 °C et 8 °C, laisser la solution diluée atteindre la température ambiante avant administration. Ne pas administrer en injection intraveineuse directe ou en bolus. Voir le tableau 5 et le tableau 6 pour des informations sur la durée minimale de perfusion. La perfusion doit être administrée à l'aide d'un filtre de 0,2 µm.
- Si le médicament n'est pas utilisé immédiatement après dilution, les durées de conservation ne doivent pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C ou 4 heures à température ambiante, en prenant en compte la durée de perfusion prévue.
Tableau 25 : Tableau de référence pour l'administration de
la dose de charge pour Ultomiris 300 mg/3 mL et 1 100 mg/11 mL solutions à
diluer pour perfusion
| Poids (kg)a |
Dose de charge (mg) |
Volume d'Ultomiris (mL) |
Volume de solution de NaCl (diluant)b (mL) |
Volume total (mL) |
| ≥ 10 à < 20 | 600 | 6 | 6 | 12 |
| ≥ 20 à < 30 | 900 | 9 | 9 | 18 |
| ≥ 30 à < 40 | 1 200 | 12 | 12 | 24 |
| ≥ 40 à < 60 | 2 400 | 24 | 24 | 48 |
| ≥ 60 à < 100 | 2 700 | 27 | 27 | 54 |
| ≥ 100 | 3 000 | 30 | 30 | 60 |
a Poids au moment du traitement.
Tableau 26 : Tableau de référence pour l'administration des doses d'entretien pour Ultomiris 300 mg/3 mL et 1 100 mg/11 mL solutions à diluer pour perfusion
| Poids (kg)a |
Dose d'entretien (mg) |
Volume d'Ultomiris (mL) |
Volume de solution de NaCl (diluant)b (mL) |
Volume total (mL) |
| ≥ 10 à < 20 | 600 | 6 | 6 | 12 |
| ≥ 20 à < 30 | 2 100 | 21 | 21 | 42 |
| ≥ 30 à < 40 | 2 700 | 27 | 27 | 54 |
| ≥ 40 à < 60 | 3 000 | 30 | 30 | 60 |
| ≥ 60 à < 100 | 3 300 | 33 | 33 | 66 |
| ≥ 100 | 3 600 | 36 | 36 | 72 |
a Poids au moment du traitement.
Tableau 27 : Tableau de référence pour l'administration des doses supplémentaires pour Ultomiris 300 mg/3 mL et 1 100 mg/11 mL solutions à diluer pour perfusion
| Poids (kg)a |
Dose supplémentaire (mg) |
Volume d'Ultomiris (mL) |
Volume de solution de NaCl (diluant)b (mL) |
Volume total (mL) |
| ≥ 40 à < 60 | 600 | 6 | 6 | 12 |
| 1 200 | 12 | 12 | 24 | |
| 1 500 | 15 | 15 | 30 | |
| ≥ 60 à < 100 | 600 | 6 | 6 | 12 |
| 1 500 | 15 | 15 | 30 | |
| 1 800 | 18 | 18 | 36 | |
| ≥ 100 | 600 | 6 | 6 | 12 |
| 1 500 | 15 | 15 | 30 | |
| 1 800 | 18 | 18 | 36 |
a Poids au moment du traitement.
b Ultomiris ne doit être dilué qu'en utilisant une solution
injectable de chlorure de sodium à 9 mg/mL (0,9 %).
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Ultomiris 300 mg/30 mL solution à diluer pour perfusion
Ce médicament doit être dilué à une concentration finale de 5 mg/mL.
Une
technique aseptique doit être utilisée.
Préparer
Ultomiris solution à diluer pour perfusion en suivant les instructions
suivantes :
- Le nombre de flacons à utiliser pour la dilution est déterminé en fonction du poids du patient et de la dose prescrite, voir la rubrique Posologie et mode d'administration.
- Avant dilution, la solution contenue dans les flacons doit être inspectée visuellement pour vérifier l'absence de particules ou de précipité. Ne pas utiliser si des particules ou un précipité sont observés.
- Le volume calculé de médicament est prélevé du nombre de flacons approprié et dilué dans une poche pour perfusion en utilisant comme diluant une solution injectable de chlorure de sodium à 9 mg/mL (0,9 %). Voir ci-dessous les tableaux de référence pour l'administration. Mélanger doucement le produit. Ne pas agiter le produit.
- Après dilution, la concentration finale de la solution à perfuser est de 5 mg/mL.
- La solution préparée doit être administrée immédiatement après la préparation, sauf si elle est conservée à une température comprise entre 2 °C et 8 °C. En cas de conservation à une température comprise entre 2 °C et 8 °C, laisser la solution diluée atteindre la température ambiante avant administration. Ne pas administrer en injection intraveineuse directe ou en bolus. Voir le tableau 7 et le tableau 8 pour des informations sur la durée minimale de perfusion lors de l'administration. La perfusion doit être administrée à l'aide d'un filtre de 0,2 µm.
- Si le médicament n'est pas utilisé immédiatement après dilution, les durées de conservation ne doivent pas dépasser 24 heures à une température comprise entre 2 °C et 8 °C ou 6 heures à température ambiante, en prenant en compte la durée de perfusion prévue.
Tableau 28 : Tableau de
référence pour l'administration de la dose de charge pour Ultomiris 300 mg/30
mL solution à diluer pour perfusion
| Poids (kg)a |
Dose de charge (mg) |
Volume d'Ultomiris (mL) |
Volume de solution de NaCl (diluant)b (mL) |
Volume total (mL) |
| ≥ 10 à < 20 | 600 | 60 | 60 | 120 |
| ≥ 20 à < 30 | 900 | 90 | 90 | 180 |
| ≥ 30 à < 40 | 1 200 | 120 | 120 | 240 |
| ≥ 40 à < 60 | 2 400 | 240 | 240 | 480 |
| ≥ 60 à < 100 | 2 700 | 270 | 270 | 540 |
| ≥ 100 | 3 000 | 300 | 300 | 600 |
a Poids au moment du traitement.
b Ultomiris ne doit être dilué qu'en utilisant une solution
injectable de chlorure de sodium à 9 mg/mL (0,9 %).
Tableau 29 : Tableau de référence pour l'administration des doses d'entretien pour
Ultomiris 300 mg/30 mL solution à diluer pour perfusion
| Poids (kg)a |
Dose d'entretien (mg) |
Volume d'Ultomiris (mL) |
Volume de solution de NaCl (diluant)b (mL) |
Volume total (mL) |
| ≥ 10 à < 20 | 600 | 60 | 60 | 120 |
| ≥ 20 à < 30 | 2 100 | 210 | 210 | 420 |
| ≥ 30 à < 40 | 2 700 | 270 | 270 | 540 |
| ≥ 40 à < 60 | 3 000 | 300 | 300 | 600 |
| ≥ 60 à < 100 | 3 300 | 330 | 330 | 660 |
| ≥ 100 | 3 600 | 360 | 360 | 720 |
a Poids au moment du traitement.
b Ultomiris ne doit être dilué qu'en utilisant une solution
injectable de chlorure de sodium à 9 mg/mL (0,9 %).
Tableau 30
: Tableau de référence pour l'administration des doses supplémentaires pour
Ultomiris 300 mg/30 mL solution à diluer pour perfusion
| Poids (kg)a |
Dose supplémentaire (mg) |
Volume d'Ultomiris (mL) |
Volume de solution de NaCl (diluant)b (mL) |
Volume total (mL) |
| ≥ 40 à < 60 | 600 | 60 | 60 | 120 |
| 1 200 | 120 | 120 | 240 | |
| 1 500 | 150 | 150 | 300 | |
| ≥ 60 à < 100 | 600 | 60 | 60 | 120 |
| 1 500 | 150 | 150 | 300 | |
| 1 800 | 180 | 180 | 360 | |
| ≥ 100 | 600 | 60 | 60 | 120 |
| 1 500 | 150 | 150 | 300 | |
| 1 800 | 180 | 180 | 360 |
a Poids au moment du traitement.
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Liste I.
Médicament nécessitant une surveillance particulière pendant le traitement.
Prescription
réservée aux spécialistes et services HEMATOLOGIE.
Prescription réservée aux spécialistes et services MEDECINE INTERNE.
Prescription réservée aux spécialistes et services NEPHROLOGIE.
Prescription réservée aux spécialistes et services NEUROLOGIE.
Prescription réservée aux spécialistes et services PEDIATRIE.
Réservé à l'usage HOSPITALIER.
Extension
d'indication (JO du 23/04/2024) :
La prise en charge des spécialités
ci-dessous est étendue aux indications suivantes :
- SHUa :
traitement du syndrome hémolytique et urémique atypique (SHUa)
chez les patients pesant 10 kg ou plus, naïfs d'inhibiteur du complément ou
ayant reçu un traitement par l'eculizumab pendant au
moins 3 mois et présentant des signes de réponse à l'eculizumab
;
- MAg : en
addition au traitement standard, incluant les immunosuppresseurs de première
ligne, chez les patients adultes atteints de myasthénie auto-immune généralisée
et présentant des anticorps anti-récepteurs de l'acétylcholine (RACh) restant symptomatiques.
En outre, compte tenu de la place
dans la stratégie thérapeutique de ULTOMIRIS (ravulizumab)
restreinte en tant que traitement de première intention en addition au
traitement standard, incluant les immunosuppresseurs de première ligne, chez
les patients adultes atteints de myasthénie auto-immune généralisée et
présentant des anticorps anti-récepteurs de l'acétylcholine (RACh) restant symptomatiques, sa prise en charge dans cette
indication est subordonnée à une validation par concertation d'une équipe
d'experts appartenant à la filière FILNEMUS de l'indication de ULTOMIRIS (ravulizumab). Une validation par une RCP pourra être
proposée à la commission « thérapie innovante » de la filière FILNEMUS dans les
situations où l'indication est plus difficile à poser.
- NMOSD : dans le traitement de la
maladie du spectre de la neuro-myélite optique chez les patients adultes
présentant des anticorps anti-aquaporine 4 (AQP4) et en échec des traitements
de fond immunosuppresseurs (rituximab, azathioprine, mycophénolatemofétil).
En outre, compte tenu de la place
dans la stratégie thérapeutique de ULTOMIRIS (ravulizumab)
restreinte et du risque d'utilisation au-delà du périmètre de remboursement
défini par la Commission sur la base des données disponibles d'efficacité et de
tolérance, sa prise en charge dans cette indication est subordonnée au suivi du
patient dans un centre de ressources et de compétences de la sclérose en
plaques ou dans un centre du réseau MIRCEM (maladies inflammatoires rares du
cerveau et de la moelle), avec une prescription restreinte aux neurologues dans
le cadre d'une RCP.
Solution à diluer pour perfusion (solution à diluer stérile).
Solution translucide, transparente à jaunâtre, de pH 7,4.
Boîte d'un flacon.
11 mL de solution à diluer stérile dans un flacon (verre de type I) muni d'un bouchon et d'un opercule.
Ultomiris est une formulation de ravulizumab, produit en culture de cellules d'ovaire de hamster chinois (CHO) par la technologie de l'ADN recombinant.
Chaque flacon de 11 mL contient 1 100 mg de ravulizumab (100 mg/mL).
Après dilution, la concentration finale de la solution à perfuser est de 50 mg/mL.
Excipient(s) à effet notoire :
Sodium (16,8 mg par flacon de 11 mL)
Pour la liste complète des excipients, voir rubrique Liste des excipients.
Phosphate de sodium dibasique heptahydraté
Phosphate de sodium monobasique monohydraté
Polysorbate 80
Arginine
Saccharose
Eau pour préparations injectables